-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2015; 5(1): 10-13
doi:10.5923/j.ajoc.20150501.02
Synthesis and Spectroscopic Properties of 1H-Cyclohepta[2,1-b:3,4-b’]diindole and Molecular Structure of its Protonated Species
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLYoshimitsu Kumai 1, Ryuta Miyatake 2, Yuka Sugeno 1, Akira Ohta 1, Mitsunori Oda 1
1Department of Chemistry, Faculty of Science, Shinshu University, Matsumoto, Nagano, Japan
2Centre of Environmental Conservation and Research Safety, University of Toyama, Toyama, Japan
Correspondence to: Mitsunori Oda , Department of Chemistry, Faculty of Science, Shinshu University, Matsumoto, Nagano, Japan.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
The title compound 9 was synthesized by condensation of 2,2’-biindole and 1-chloro-3-(N,N-dimethylamino) propenium chloride in 1,2-dichloroethane. NMR and UV-vis absorption spectra of 9 under neutral, acidic and basic conditions were discussed in respect of its protonation and deprotonation. A crystal structure of its protonated species, 9H+TfO–, was determined by X-ray diffraction analysis, indicating its planar and symmetrical structure. Change of absorption spectra of 9 in the presence of a large excess of various metal ions was studied to find out a mode of interaction between 9 and metal ion.
Keywords: Biindole, Azaazulene, Azepine, X-ray structure, Absorption spectra, DFT calculations
Cite this paper: Yoshimitsu Kumai , Ryuta Miyatake , Yuka Sugeno , Akira Ohta , Mitsunori Oda , Synthesis and Spectroscopic Properties of 1H-Cyclohepta[2,1-b:3,4-b’]diindole and Molecular Structure of its Protonated Species, American Journal of Organic Chemistry, Vol. 5 No. 1, 2015, pp. 10-13. doi: 10.5923/j.ajoc.20150501.02.
Article Outline
1. Introduction
- There were reported two marine natural products having two indole units around a fully unsaturated seven-membered ring, iheyamine A (1) [1] and cauresin (2) [2–5], the former of which exhibits cytotoxic activity against tumor cells. [1] In respect to their unique structures and biological activity, various compounds (3–8) structurally related to 1 and 2 have been synthesized (Figure 1). [3–6]
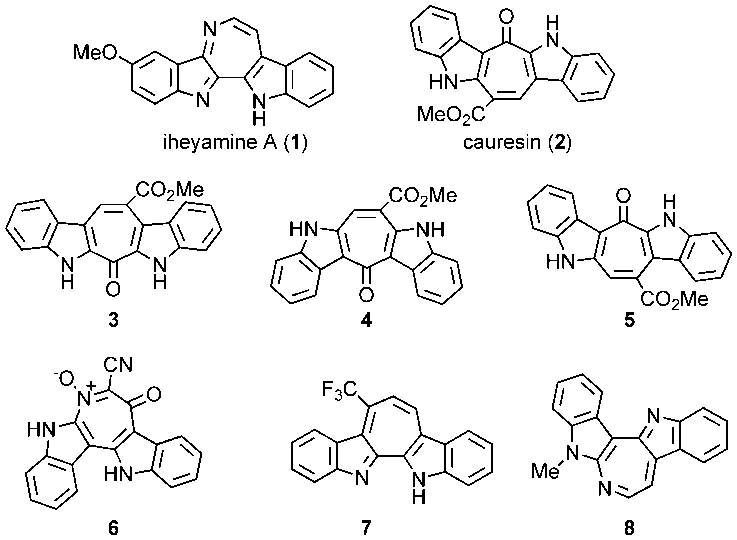 | Figure 1. Marine natural products (1–2) having two indole units and structurally related synthetic compounds (3–8) |
 systems have not been reported yet. Herein we describe synthesis of the title compound 9, 1H-cyclohepta[2,1-b:3,4-b’]diindole, which is the parent compound for 7, its absorption properties under various conditions, and also X-ray structure of its trifluoromethane- sulfonic acid (TfOH) salt.
systems have not been reported yet. Herein we describe synthesis of the title compound 9, 1H-cyclohepta[2,1-b:3,4-b’]diindole, which is the parent compound for 7, its absorption properties under various conditions, and also X-ray structure of its trifluoromethane- sulfonic acid (TfOH) salt.2. Results and Discussion
2.1. Synthesis and properties of 1H-cyclohepta- [2,1-b:3,4-b’]diindole (9)
- The title compound 9 was synthesized, in a similar way of Baraznenok et al., [6] by condensation of 2,2’-biindole (10) [9] with 1-chloro-3-(N,N-dimethylamino)propenium chloride (11), which was generated in situ by reaction of 3-dimethylaminoacrolein with oxalyl chloride (Scheme 1) [10].
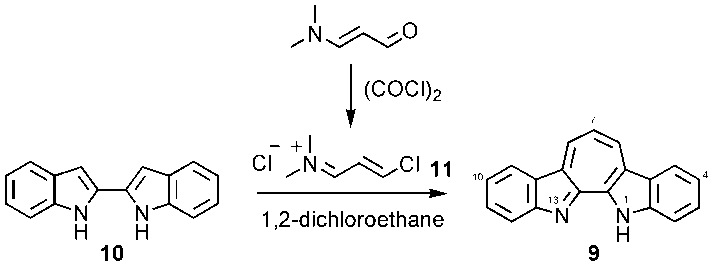 | Scheme 1. Synthesis of 9 |
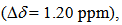 as seen in azulenes and azaazulenes, which have alternatively fluctuated
as seen in azulenes and azaazulenes, which have alternatively fluctuated 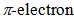 density around their peripheral ring frameworks [11, 12].The UV-vis spectrum of 9 in CH3CN exhibits mainly two absorption bands, strong band around 330 nm and weak band at 495 nm (Figure 2). In an acidic medium of trifluoroacetic acid (TFA), the long wavelength absorption exhibits a blue shift by 35 nm and color of the solution turns to yellow orange. On the other hand, in a basic medium of 16%.
density around their peripheral ring frameworks [11, 12].The UV-vis spectrum of 9 in CH3CN exhibits mainly two absorption bands, strong band around 330 nm and weak band at 495 nm (Figure 2). In an acidic medium of trifluoroacetic acid (TFA), the long wavelength absorption exhibits a blue shift by 35 nm and color of the solution turns to yellow orange. On the other hand, in a basic medium of 16%.  | Figure 2. Absorption spectra of 9 in CH3CN (green), TFA (red), and NaOH aq./DMSO (blue). Inset is expanded spectra at a range of 280–380 nm |
 8.25 ppm) are deshielded and those of 12 (
8.25 ppm) are deshielded and those of 12 (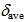 7.91 ppm) slightly shielded compared with that of 9 (
7.91 ppm) slightly shielded compared with that of 9 ( 7.93 ppm) in CDCl3.
7.93 ppm) in CDCl3. | Scheme 2. Protonation and deprotonation of 9 |
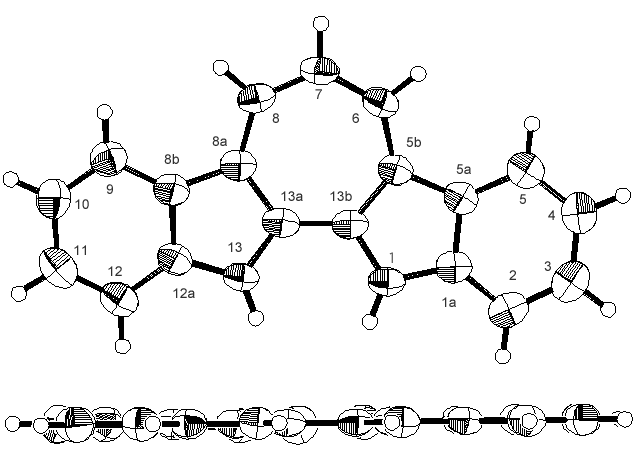 | Figure 3. ORTEP drawings of 9H+TfO–, one of two different molecules. Top views (top) and side view (bottom). The counter anion (TfO–) was omitted |
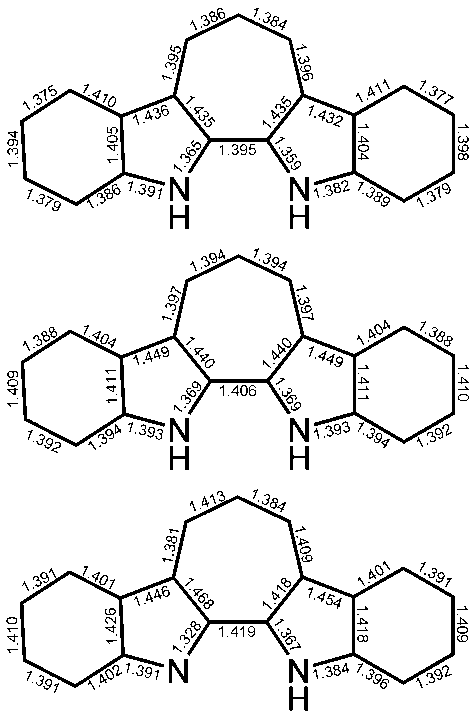 | Figure 4. Average bond lengths (in Å) of the crystal structures of 9H+TfO– (top) and bond lengths of the calculated structures of 9H+ (middle) and 9 (bottom) |
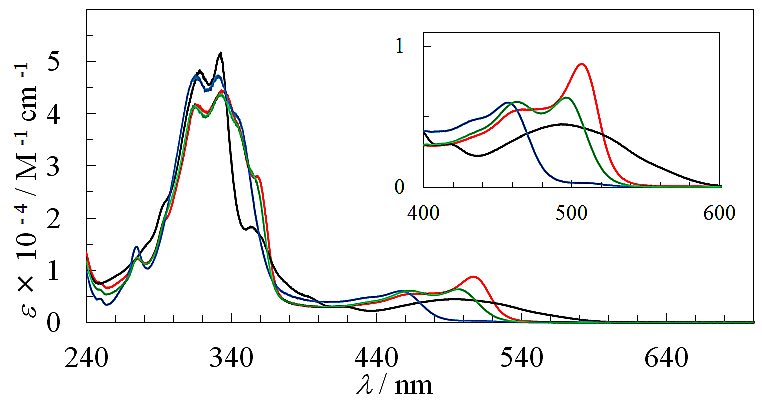 | Figure 5. Absorption spectra of 9 in the presence of a large excess (100 eq.) of Li+ (black), Mg2+ (blue), Zn2+ (green), and Cd2+ (red). Inset is expanded spectra at a range of 400–600 nm |
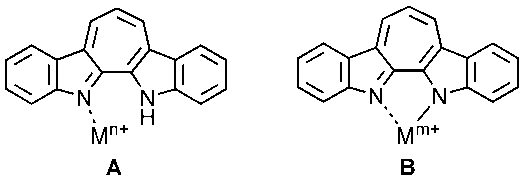 | Figure 6. Modes of complexation of 9 with metal ions |
3. Experiments
3.1. General
- Melting points were measured on a Yanaco MP-3 and are uncorrected. IR spectra were recorded on a JASCO FT/IR-4100 spectrometer. UV-vis spectra were measured on a Shimadzu UV-2550 spectrometer. 1H- and 13C-NMR spectra were recorded on JEOL
 400 and ECA500 spectrometers. Chemical shift value of tetramethylsilane (
400 and ECA500 spectrometers. Chemical shift value of tetramethylsilane (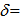 0 ppm) for 1H-NMR and 13C-NMR spectra was used as internal standard. Mass spectra were measured on a JMS-700 mass spectrometer. Column chromatography was performed with Silica gel 60N from Kanto Chem. Co. 1,2-Dichloro- ethane was purchased from Kanto Chem. Co. and was distilled over CaH2. Oxalyl chloride was purchased from Wako Chem. Co. and was used without purification. TfOH and TFA were purchased from Tokyo Chem. Industry, Inc. 2,2’-Biindole was prepared in two steps from o-anisidine according to a procedure reported by Bergman et al. [9]
0 ppm) for 1H-NMR and 13C-NMR spectra was used as internal standard. Mass spectra were measured on a JMS-700 mass spectrometer. Column chromatography was performed with Silica gel 60N from Kanto Chem. Co. 1,2-Dichloro- ethane was purchased from Kanto Chem. Co. and was distilled over CaH2. Oxalyl chloride was purchased from Wako Chem. Co. and was used without purification. TfOH and TFA were purchased from Tokyo Chem. Industry, Inc. 2,2’-Biindole was prepared in two steps from o-anisidine according to a procedure reported by Bergman et al. [9]3.2. 1H-Cyclohepta[2,1-b;3,4-b’]diindole (9)
- A solution of 119 mg (1.20 mmol) of N,N-dimethylamino-acrolein in 3 mL of 1,2-dichloroethane (DCE) was added to an ice-cooled solution of 151 mg (1.00 mmol) of oxalyl- chloride in 2 mL of DCE. To this mixture was added a suspension of 232 mg (1.00 mmol) of 2,2’-biindole (10) in 15 mL of DCE and the mixture was refluxed on an oil bath for 16 h under nitrogen atmosphere. The resulted bright yellow reaction mixture was poured into a saturated Na2CO3 aqueous solution and the water layer was extracted with chloroform (50 mL x 3). The combined organic layer was washed with a brine and was dried over Na2SO4. The solvent was removed and the residue was purified by silica gel column chromatography eluted with 5%EtOH-chloroform to give 154 mg (57% yield) of 9 as dark red plates, m.p. = 297–298℃. 1H NMR (400 MHz, CDCl3)
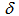 = 4.72 (br. 1H, N–H), 7.46 (t, J = 7.7 Hz, 2H, H-4,10), 7.56 (t, J = 7.7 Hz, 2H, H-3,11), 7.64 (d, J = 7.7 Hz, 2H, H-2,12), 7.70 (t, J = 10.0 Hz, 1H, H-7), 8.36 (d, J = 7.7 Hz, 2H, H-5,9), 8.90 (d, J = 10.0 Hz, 2H, H-6,8) ppm; 1H NMR (400 MHz, TFA-d)
= 4.72 (br. 1H, N–H), 7.46 (t, J = 7.7 Hz, 2H, H-4,10), 7.56 (t, J = 7.7 Hz, 2H, H-3,11), 7.64 (d, J = 7.7 Hz, 2H, H-2,12), 7.70 (t, J = 10.0 Hz, 1H, H-7), 8.36 (d, J = 7.7 Hz, 2H, H-5,9), 8.90 (d, J = 10.0 Hz, 2H, H-6,8) ppm; 1H NMR (400 MHz, TFA-d) 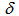 =7.67 (t, J = 7.3 Hz, 2H, H-4,10), 7.87 (t, J = 7.3 Hz, 2H, H-3,11), 7.90 (d, J = 7.3 Hz, 2H, H-2,12), 8.29 (t, J = 9.9 Hz, 1H, H-7), 8.42 (d, J = 7.3 Hz, 2H, H-5,9), 9.32 (d, J = 9.9 Hz, 2H, H-6,8) ppm; 1H NMR (400MHz, 16% NaOD- D2O/DMSO-d6 = 1 : 0.6)
=7.67 (t, J = 7.3 Hz, 2H, H-4,10), 7.87 (t, J = 7.3 Hz, 2H, H-3,11), 7.90 (d, J = 7.3 Hz, 2H, H-2,12), 8.29 (t, J = 9.9 Hz, 1H, H-7), 8.42 (d, J = 7.3 Hz, 2H, H-5,9), 9.32 (d, J = 9.9 Hz, 2H, H-6,8) ppm; 1H NMR (400MHz, 16% NaOD- D2O/DMSO-d6 = 1 : 0.6) 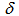 = 7.36 (t, J = 7.8 Hz, 2, H-4,10), 7.38 (t, J = 9.5 Hz, 1H, H-7), 7.58 (t, J = 7.8 Hz, 2H, H-3,11), 7.95 (d, J = 7.8 Hz, 2H, H-2,12), 8.33 (d, J = 7.8 Hz, 2H, H-5,9), 8.84 (d, J = 9.5 Hz, 2H, H-6,8) ppm; 13C NMR (100 MHz, CDCl3)
= 7.36 (t, J = 7.8 Hz, 2, H-4,10), 7.38 (t, J = 9.5 Hz, 1H, H-7), 7.58 (t, J = 7.8 Hz, 2H, H-3,11), 7.95 (d, J = 7.8 Hz, 2H, H-2,12), 8.33 (d, J = 7.8 Hz, 2H, H-5,9), 8.84 (d, J = 9.5 Hz, 2H, H-6,8) ppm; 13C NMR (100 MHz, CDCl3) 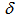 =115.4 (C-2,12), 120.3 (C-5,9), 122.1 (C-7), 122.4 (C-4,10), 126.3 (C-5a,8b), 129.3 (C-8,11), 131.1 (C-5b,8a), 132.4 (C-6,8), 142.0 (C-13a,13b), 144.5 (C-1a, 12a) ppm; IR (KBr)
=115.4 (C-2,12), 120.3 (C-5,9), 122.1 (C-7), 122.4 (C-4,10), 126.3 (C-5a,8b), 129.3 (C-8,11), 131.1 (C-5b,8a), 132.4 (C-6,8), 142.0 (C-13a,13b), 144.5 (C-1a, 12a) ppm; IR (KBr) 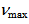 = 1402vs, 1393vs, 1215s, 1203s, 732s cm-1; UV-vis (CH3CN)
= 1402vs, 1393vs, 1215s, 1203s, 732s cm-1; UV-vis (CH3CN) 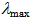 =
= 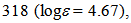 332 (4.70), 353 (4.25), 394sh (3.69), 419 (3.45), 494 (3.65) nm; MS (70 eV) m/z (rel int.) = 268 (M+, 100), 267 (13), 266 (11), 240 (4), 134 (10). HRMS Calcd for C19H12N2 268.1001; Found 268.1001.
332 (4.70), 353 (4.25), 394sh (3.69), 419 (3.45), 494 (3.65) nm; MS (70 eV) m/z (rel int.) = 268 (M+, 100), 267 (13), 266 (11), 240 (4), 134 (10). HRMS Calcd for C19H12N2 268.1001; Found 268.1001.3.3. 1,13H-Cyclohepta[2,1-b;3,4-b’]diindolium triflate (9H+TfO–)
- To a solution of 11.2 mg
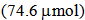 of TfOH in 10 mL of dichloromethane was added 20.0 mg (74.6 mol) of 9. The solvent was removed under reduced pressure and the residue was recrystallized from a mixture of dichloromethane and hexane to give 27.0 mg (88% yield) of 9H+TfO– as yellow solids, m.p. > 300℃. 1H NMR (500 MHz, CDCl3)
of TfOH in 10 mL of dichloromethane was added 20.0 mg (74.6 mol) of 9. The solvent was removed under reduced pressure and the residue was recrystallized from a mixture of dichloromethane and hexane to give 27.0 mg (88% yield) of 9H+TfO– as yellow solids, m.p. > 300℃. 1H NMR (500 MHz, CDCl3) 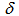 = 1.51 (s, 2H, N–H), 7.60 (t, J = 7.9 Hz, 2H, H-4,10), 7.83 (t, J = 7.9 Hz, 2H, H-3,11), 8.05 (d, J = 7.9 Hz, 2H, H-2,12), 8.23 (t, J = 10.0 Hz, 1H, H-7), 8.38 (d, J = 7.9 Hz, 2H, H-5,9), 9.27 (d, J = 10.0 Hz, 2H, H-6,8) ppm; 13C NMR (126 MHz, CDCl3)
= 1.51 (s, 2H, N–H), 7.60 (t, J = 7.9 Hz, 2H, H-4,10), 7.83 (t, J = 7.9 Hz, 2H, H-3,11), 8.05 (d, J = 7.9 Hz, 2H, H-2,12), 8.23 (t, J = 10.0 Hz, 1H, H-7), 8.38 (d, J = 7.9 Hz, 2H, H-5,9), 9.27 (d, J = 10.0 Hz, 2H, H-6,8) ppm; 13C NMR (126 MHz, CDCl3) 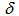 = 114.5, 120.7, 124.2, 124.7, 126.2, 131.7, 132.5, 135.1, 136.9, 141.2ppm [15]; IR (KBr)
= 114.5, 120.7, 124.2, 124.7, 126.2, 131.7, 132.5, 135.1, 136.9, 141.2ppm [15]; IR (KBr) 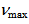 = 1620s, 1397vs, 1295vs, 1254s, 1210vs, 1166vs, 1027vs, 749vs, 731s, 635s cm-1. A sample for X-Ray crystallographic analysis was obtained by further recrystallization from methanol.
= 1620s, 1397vs, 1295vs, 1254s, 1210vs, 1166vs, 1027vs, 749vs, 731s, 635s cm-1. A sample for X-Ray crystallographic analysis was obtained by further recrystallization from methanol.3.4. X-Ray Crystallographic Analysis of 9H+TfO–
- Diffraction measurements were conducted using a Rigaku R-AXIS RAPID diffractometer at –100℃. Crystal data for 9H+TfO– are as follows; triclinic, space group; P-1 (# 2), a; 7.0362(2) Å, b; 12.5278(3) Å, c; 20.7784(4) Å,
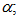 96.7827(7)°,
96.7827(7)°, 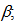 90.0115(7)°,
90.0115(7)°, 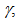 90.0034(7)°,
90.0034(7)°, 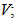 1818.76(6) Å3, Z; 4, R; 0.0893, wR2; 0.2353, R1; 0.0729
1818.76(6) Å3, Z; 4, R; 0.0893, wR2; 0.2353, R1; 0.0729 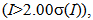 and S; 1.092. The relatively large R values are mainly attributed to thermal vibration of fluorine atoms of the triflate anion. Tables of fractional atomic coordinates, thermal parameters, bond lengths, and angles have been deposited at the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, United Kingdom (CCDC 1038190) [Derect line: +44 1223 762910, Fax: +44 (0) 1233 336033, e-mail: deposit@ccdc.cam.ac.uk].
and S; 1.092. The relatively large R values are mainly attributed to thermal vibration of fluorine atoms of the triflate anion. Tables of fractional atomic coordinates, thermal parameters, bond lengths, and angles have been deposited at the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, United Kingdom (CCDC 1038190) [Derect line: +44 1223 762910, Fax: +44 (0) 1233 336033, e-mail: deposit@ccdc.cam.ac.uk].4. Conclusions
- We have demonstrated that the title parent compound 9 could be synthesized from 2,2’-biindole (10) by its condensation with1-(N,N-dimethylamino)-3-chloropropenium chloride. Compound 9 shows not only basic nature but also acidic nature. Spectral changes of UV-vis and NMR spectra were discussed based on protonation and deprotonation of 9. Changes of the UV-vis spectrum in the presence of various metal ions were also described.
ACKNOWLEDGEMENTS
- Financial support from the Faculty of Science in Shinshu University (for M.O.) is greatly acknowledged.