-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2015; 5(1): 1-9
doi:10.5923/j.ajoc.20150501.01
Synthesis and Antimicrobial Evaluation of New Polyfunctionally Substituted Heterocyclic Compounds Derived from 2-cyano-N-(3-cyanoquinolin-2-yl)acetamide
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLKamal M. El-Gaml
Organic Chemistry Department, Faculty of Pharmacy, Al-Azhar University, Nasr City, Cairo, Egypt
Correspondence to: Kamal M. El-Gaml, Organic Chemistry Department, Faculty of Pharmacy, Al-Azhar University, Nasr City, Cairo, Egypt.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
New 2-cyano-N-(3-cyanoquinolin-2-yl) acetamide (3) was utilized as key intermediate for the synthesis of some new thiazole, pyrazole, thiophene, oxazine and pyridazine derivatives. Newly synthesized Pyrazole derivatives were obtained by diazotization of 3 with the desired diazonium chloride that gave the hydrazine derivatives 4a–e. Also, the reactivity of the hydrazine towards hydrazine hydrate to give Pyrazole derivatives was studied. Furthermore, treatment of 3with elemental sulfur and phenyl isothiocyanate or malononitrile afford thiazole and thiophene derivatives respectively. When 3react with phenyl isothiocyanate and KOH afforded the intermediate salt which reacted in situ with chloroacetyl chloride or bromoacetone to give thiazole derivatives 8 and 9. In addition, when 3 react with phenacyl bromide it gives 10 that can be utilized for synthesis of pyridazine and oxazine derivatives. All the newly synthesized compounds were screened for their antimicrobial activity and some of the synthesized derivatives showed marked antimicrobial but other showed moderate antimicrobial activity.
Keywords: N-quinolinyl-cyanoactamide, Thiazole, Pyrazole, Thiophene, Oxazine, Pyridazine, Antimicrobial activity
Cite this paper: Kamal M. El-Gaml, Synthesis and Antimicrobial Evaluation of New Polyfunctionally Substituted Heterocyclic Compounds Derived from 2-cyano-N-(3-cyanoquinolin-2-yl)acetamide, American Journal of Organic Chemistry, Vol. 5 No. 1, 2015, pp. 1-9. doi: 10.5923/j.ajoc.20150501.01.
Article Outline
1. Introduction
- Cyanoquinoline nucleus is often used for the design of many synthetic compounds with diverse pharmacological properties [1] in addition; to cyanoactamide are highly reactive moieties because it extensively used as reactants or reaction intermediates due to the carbonyl and the cyano functions of these compounds are suitable to form a variety of heterocyclic compounds. The synthesis of cyanoactamide may be carried out in several ways. The most economical method involves the treatment of substituted aryl or heteryl amines with alkyl cyanoacetate using different reaction conditions to yield cyanoactamide derivatives [2]. Cyanoactamide and their related heterocyclic derivatives have generated a great deal of attention due to their interesting pharmaceutical activities include antimicrobial [3], antifungal [4], insulin releasing [5], carbonic anhydrase inhibitory [6], anti-inflammatory [7], and antitumor properties [8]. Due to chemical properties of these compounds because it possessing both electrophilic and nucleophilic moieties make it useful for synthesis of three membered rings [9], five member rings such as pyrrole [10] and pyrazole and their fused derivatives [11] in addition to, imidazole [12], thiazole [13], 1,3-dithiolane [14], thiadiazole and their derivatives [15, 16], also, six membered rings such as pyridine, [17] pyrane, [18] pyradazine, [19] thiazine, [20] and triazine derivatives [21].
2. Experimental
2.1. General
- Melting points were measured in capillary tube on a Graffin melting point apparatus and are uncorrected. The IR spectra were recorded on PyeUnicam SP 1000 IR spectrophotometer using KBr discs (λmax in cm-1). 1HNMR spectra were performed either on a Jeol ECA (500 MHz) or Gemini 300BB (300MHz) spectrometer, using TMS as internal standard and DMSO-d6 as solvent; the chemical shifts are reported in ppm (δ) and coupling constant (J) values are given in Hertz (Hz). Signal multiplicities are represented by s (singlet), d (doublet), t (triplet), q (quadruplet), and m (multiplet). All of the new compounds were analyzed for C, H and N and agreed with the proposed structures within ±0.4% of the theoretical values by the automated CHN analyzer. Mass spectra were recorded on Hewlett Packard 5988 spectrometer at the RCMB. The purity of the compounds was checked by thin layer chromatography (TLC) on Merck silica gel 60 F254 precoated sheets. All analyses were performed at the Micro analytical Center Unit of Cairo University, Cairo, Egypt. The starting Compounds 1 were prepared according to reported procedures [22-25].
2.2. Chemistry
2.2.1. 2-Aminoquinoline-3-carbonitrile (2) [26-29]
- To the well stirred solution of 2-chloroquinoline-3- carbonitrile (1) [22-25] (1.88g, 0.002 mole) and tetrabutylammonium bromide (0.0005 mole, 0.202g) in chlorobenzene (15ml) was added sodium azide (0.006 mole, 0.390) in water 5ml, the reaction mixture was stirred under reflux for 1.5 hr, at completion of time powdered sodium borohydride (0.008mole, 0.302g) was added to the reaction mixture portion wise cautiously over a period of 30 mint. The same reaction mixture was then refluxed for 1-2 hrs. On completion (TLC) the aqueous phase was separated and extracted with chlorobenzene, and combined organic layer was washed with water and dried with anhydrous Na2SO4. The solvent was recovered in vacuum, the content was treated with n-hexane and the solid thus formed was filtered, washed with cold methanol, dried and crystalized from methanol : chloroform (7:3 V/V) .Our procedure differ from the reported [26-29] one and it modify the other one [29] to get the compound 2.Yield 75%, M. p. 228 – 230℃. IR (KBr) cm-1: 3350, 3296, 2972, 2865, 2215. 1HNMR (DMSO-d6) δ: 6.54(s, 2H, NH2), 7.19-7.23 (t, 1H, C6-H quinoline), 7.42-7.46 (t, 1H, C7-H quinoline), 7.59-6.62 (d, 1H, C5-H quinoline), 7.76-7.80 (d, 1H, C8-H quinoline), 8.36 (s, 1H, C4-H quinoline). 13C NMR (DMSO-d6) δ: 84.6 (C-3), 117.7 (CN), 119.4, 122.3, 125.1, 127.2, 132.7 (Ar-C), 153.9 (C-8), 156.8 (C-4), 164.1 (C-2). MS (m/z): 183.21 (53, M+). Anal. Calc. For C10H7N3: C, 70.99; H, 4.17; N, 24.84. Found. C, 71.10; H, 4.44; N, 24.91.
2.2.2. 2-Cyano-N-(3-cyanoquinolin-2-yl) acetamide (3)
- To a solution of 2-aminoquinoline-3-carbonitrile (2) (2.36 g, 0.01 mol) in dimethylformamide (30 ml), ethyl cyanoacetate (1.13 g, 0.01 mol) was added. The reaction mixture was heated under reflux for 5 h. The solid product formed upon pouring onto ice/water mixture was collected by filtration and crystallized from 1, 4-dioxane.Yield 71%, M. p. 129 – 130℃. IR (KBr) cm-1: 3332, 3020, 2837, 2265, 2195(2CN), 1655. 1HNMR (DMSO-d6) δ: 4.28 (s, 2H, CH2), 6.83 (s, 1H, NH), 7.19-7.23 (t, 1H, C6-H quinoline), 7.55-7.58 (t, 1H, C7-H quinoline), 7.62-7.65 (d, 1H, C5-H quinoline), 7.73-7.77 (d, 1H, C8-H quinoline), 8.27 (s, 1H, C4-H quinoline). MS (m/z): 236.23 (43.15, M+). Anal. Calc. For C13H8N4O: C, 66.10; H, 3.41; N, 23.72. Found. C, 65.89; H, 3.57; N, 23.66.
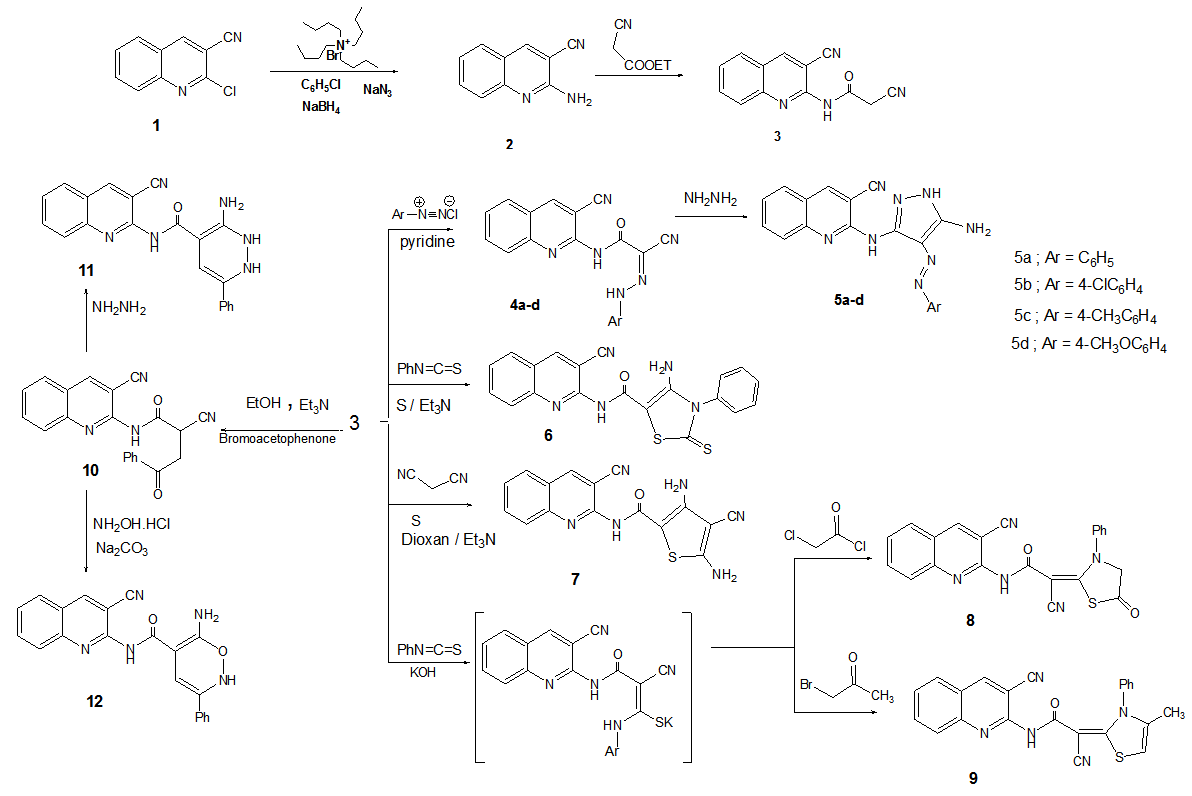 | Scheme 1. Synthesis of the precursor 2 and its cyclized products |
2.2.3. General procedure for Coupling of 2-cyano-N-(3-cyanoquinolin-2-yl) acetamide (3) with the appropriate Diazonium Salt of aromatic amines
- To a cold solution of compound 3(2.36 g, 5 mmol) in pyridine (20 mL), was added the appropriate diazonium salt of aromatic amine (aniline, 4-chloroaniline or 4-methylaniline or 4-methoxyaniline) (5 mmol) [prepared according to literature procedures] [30]. The addition was carried out portion wise with stirring at 0–5°C over a period of 30 min. After complete addition, the reaction mixture was stirred for a further 4 h then kept in an ice chest for 12 h and finally diluted with water. The precipitated solid was collected by filtration, washed with water, dried and finally recrystallized from the proper solvent to afford the corresponding couplingproducts 4a–d.2-Cyano-N-(3-cyanoquinolin-2-yl)-2-(2-phenylhydrazono) acetamide (4a)Yield (85%), mp> 300°C (from dioxane) ); IR (KBr) cm-1: 3225, 3202 (2NH), 2236, 2210(2C≡N), 2942,2852, 1650 ; 1H-NMR (DMSO-D2O): δ 6.78-701 (m, 5H, ArH), 7.23-7.25 (t, 1H, C6-H quinoline),7.66-7.69(t, 1H, C7-H quinoline), 7.75-7.77 (d, 1H, C5-H quinoline), 7.84-7.86 (d, 1H, C8-H quinoline), 8.59 (s, 1H, C4-H quinoline),10.18 (s, 1H, NH D2O exchangeable), 11.83 (s, 1H, NH, D2O exchangeable); MS (m/z): 340.34 (28.14, M+). Anal. Calc. For C19H12N6O: C, 67.05; H, 3.55; N, 24.69.Found.C, 67.11; H, 3.70; N, 24.94.2-(2-(4-Chlorophenyl) hydrazono)-2-cyano-N-(3-cyanoquinolin-2-yl) acetamide (4b)Yield (95%), mp> 300 °C (from dioxane) ); IR (KBr) cm-1: 3231, 3205 (2NH), 2242, 2219(2C≡N), 2947,2860, 1665 (C=O); 1HNMR (DMSO- D2O): δ 6.93 (d, 2H, J = 8 Hz, ArH), 7.05 (d, 2H, J = 8 Hz, ArH), 7.25-7.28 (t, 1H, C6-H quinoline), 7.56-7.59(t, 1H, C7-H quinoline), 7.64-7.67 (d, 1H, C5-H quinoline), 7.82-7.84 (d, 1H, C8-H quinoline), 8.52 (s, 1H, C4-H quinoline), 10.22 (s, 1H, NH, D2O exchangeable), 12.04 (s, 1H, NH, D2O exchangeable); MS (m/z): 374.07 (27.01, M+), 376 (9.06, M+2). Anal. Calc. For C19H11ClN6O: C, 60.89; H, 2.96; N, 22.42.Found.C, 60.76; H, 2.90; N, 22.51.2-Cyano-N-(3-cyanoquinolin-2-yl)-2-(2-p-tolylhydrazono) acetamide (4c)Yield (85%), mp 258-260°C (from ethanol); IR (KBr) cm-1: 3222, 3190 (2NH), 2235, 2214(2C≡N), 2944,2854, 1653 (C=O); 1H-NMR (DMSO- D2O): δ 2.32 (s, 3H, CH3), 6.51 (d, 2H, J = 8 Hz, ArH), 6.95 (d, 2H, J = 8 Hz, ArH), 7.22-7.26 (t, 1H, C6-H quinoline), 7.52-7.55(t, 1H, C7-H quinoline), 7.60-7.63 (d, 1H, C5-H quinoline), 7.78-7.80 (d, 1H, C8-H quinoline), 8.45 (s, 1H, C4-H quinoline), 10.42 (s, 1H, NH, D2O exchangeable), 12.02 (s, 1H, NH, D2O exchangeable); MS (m/z): 354.36 (15.07, M+). Anal. Calc. For C20H14N6O: C, 67.79; H, 3.98; N, 23.72. Found. C, 67.90; H, 3.80; N, 23.73.2-Cyano-N-(3-cyanoquinolin-2-yl)-2-(2-(4-methoxyphenyl)hydrazono) acetamide (4d)Yield (90%), mp 261-262°C (from dioxane); IR (KBr) cm-1: 3227, 3193 (2NH), 2432,2202 (2C≡N),1661 (C=O); 1H-NMR (DMSO- D2O): δ 3.71 (s, 3H, OCH3), 6.59 (d, 2H, J = 9 Hz, ArH), 6.95 (d, 2H, J = 9 Hz, ArH), 7.28-7.30 (t, 1H, C6-H quinoline),7.50-7.53(t, 1H, C7-H quinoline), 7.61-7.63 (d, 1H, C5-H quinoline), 7.76-7.78 (d, 1H, C8-H quinoline), 8.56 (s, 1H, C4-H quinoline),10.12 (s, 1H, NH, D2O exchangeable), 12.88 (s, 1H, NH, D2O exchangeable); MS (m/z): 370.36 (25.10, M+). Anal. Calc. For C20H14N6O2: C, 64.86; H, 3.81; N, 22.69. Found.C, 64.63; H, 3.88; N, 22.52.
2.2.4. General Procedure for Synthesis of Aminopyrazole Containing Compounds 5a-d
- To a solution of the compound 4a-d (5 mmol) in dioxane (20 ml), hydrazine hydrate (80%,1.0 mL, 5 mmol) was added and the reaction mixture was refluxed for 6 h and allowed to cool.The solid product obtained was filtered, washed with ethanol and dried. Recrystallization from dioxane to afforded 5a-d.2-{5-Amino-4-[phenyldiazenyl]-1H-pyrazol-3-yl} aminoquinoline-3-carbonitrile (5a)Yield (65%), mp 277-278 °C (from ethanol); IR (KBr) cm-1: 3370, 3312, 3212 (2NH, NH2), 2249(CN), 2965, 2845; 1HNMR (DMSO- D2O): δ 5.22 (s, 2H, NH2, D2O exchangeable), 7.24 (m, 5H, Ar), 7.20-7.22 (t, 1H, C6-H quinoline),7.46-7.49(t, 1H, C7-H quinoline), 7.58-7.60 (d, 1H, C5-H quinoline), 7.71-7.73 (d, 1H, C8-H quinoline), 8.22 (s, 1H, C4-H quinoline),9.77 (s, 1H, NH, D2O exchangeable), 12.98 (s, 1H, NH, D2O exchangeable); MS (m/z): 354.39 (23.04, M+). Anal. Calc. For C19H14N8: C, 64.40; H, 3.98; N, 31.62.Found.C, 64.51; H, 4.10; N, 31.44.2-{5-Amino-4-[(4-chlorophenyl) diazenyl]-1H-pyrazol-3-yl} aminoquinoline-3-carbonitrile (5b)Yield (65%), mp 290-291°C (from dioxane); IR (KBr) cm-1: 3439, 3325, 3217 (2NH, NH2), 2233(CN), 2993, 2829; 1HNMR (DMSO- D2O): δ 5.18 (s, 2H, NH2, D2O exchangeable), 6.99 (d, 2H, J = 10 Hz), 7.18 (d, 2H, J = 10 Hz), 7.26-7.29 (t, 1H, C6-H quinoline),7.52-7.55(t, 1H, C7-H quinoline), 7.66-7.68 (d, 1H, C5-H quinoline), 7.79-7.81 (d, 1H, C8-H quinoline), 8.33 (s, 1H, C4-H quinoline),9.74 (s, 1H, NH, D2O exchangeable), 13.01 (s, 1H, NHM D2O exchangeable); MS (m/z): 388.81 (14.30, M+), 390(4.90, M+2). Anal. Calc. For C19H13ClN8: C, 58.69; H, 3.37; N, 28.82.Found.C, 58.77; H, 3.59; N, 28.93.2-{5-Amino-4-[(4-methylphenyl)diazenyl]-1H-pyrazol-3-yl}aminoquinoline-3-carbonitrile (5c) Yield (65%), mp 230-232 °C (from dioxane); IR (KBr) cm-1: 3430, 3335, 3210 (2NH, NH2), 2241(CN), 2967, 2832; 1HNMR (DMSO- D2O): δ 2.21 (s, 3H, CH3), 5.16 (s, 2H, NH2, D2O exchangeable), 6.84 (d, 2H, J = 10.2 Hz), 7.10 (d, 2H, J = 10.2 Hz), 7.23-7.26 (t, 1H, C6-H quinoline), 7.50-7.53(t, 1H, C7-H quinoline), 7.63-7.66 (d, 1H, C5-H quinoline), 7.73-7.77 (d, 1H, C8-H quinoline), 8.40 (s, 1H, C4-H quinoline), 9.81 (s, 1H, NH, D2O exchangeable), 13.22 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 22.3(CH3), 78.9 (C-4-pyrazole), 96.3 (C-3-quinoline), 117.8 (CN), 119.1, 123.5, 126.9, 127.3, 128.1, 128.4, 128.4, 129.6, 129.6, 130.2, 132.9, 146.2, 148.2, 154.5(C-5-pyrazole), 154.8(C-3-pyrazole) 166.4 (C-2-quinoline).MS (m/z): 368.39 (19.12, M+). Anal. Calc. For C20H16N8: C, 65.21; H, 4.38; N, 30.42.Found.C, 65.40; H, 4.33; N, 30.19.2-{5-Amino-4-[(4-methoxyphenyl)diazenyl]-1H-pyrazol-3-yl}aminoquinoline-3-carbonitrile (5d)Yield (75%), mp 210-211°C (from dioxane); IR (KBr) cm-1: 3432, 3342, 3233 (2NH, NH2), 2234(CN), 2956, 2837; 1HNMR (DMSO- D2O): δ 3.60 (s, 3H, OCH3), 5.03 (s, 2H, NH2, D2O exchangeable), 7.00 (d, 2H, J = 9.6 Hz), 7.25 (d, 2H, J = 9.6 Hz), 7.26-7.29 (t, 1H, C6-H quinoline), 7.52-7.54(t, 1H, C7-H quinoline), 7.66-7.68 (d, 1H, C5-H quinoline), 7.78-7.80 (d, 1H, C8-H quinoline), 8.34 (s, 1H, C4-H quinoline), 9.72 (s, 1H, NH, D2O exchangeable), 13.11 (s, 1H, NH, D2O-exchangeable); 13C NMR (DMSO-d6) δ:52.1(OCH3), 80.2 (C-4-pyrazole), 95.1 (C-3-quinoline), 112.4, 112.4,118.2, 119.4 (CN), 120.4, 122.9, 125.4, 126.1, 127.2, 128.2, 132.7, 134.5 144.5, 146.3, 153.8(C-5-pyrazole), 155.1(C-3-pyrazole), 168.2 (C-2-quinoline). MS (m/z): 384.39 (19.12, M+). Anal. Calc. For C20H16N8O: C, 62.49; H, 4.20; N, 29.15.Found.C, 62.60; H, 4.33; N, 29.19.
2.2.5. 4-Amino-N-(3-cyanoquinolin-2-yl)-3-phenyl-2-thioxo-2,3-dihydrothiaz-ole-5-carboxamide (6)
- To a solution of compound3 (2.36 g, 5 mmol) in DMF containing triethylamine (1 ml), elemental sulfur (0.16 g, 5 mmol) and phenyl isothiocyanate (0.68 mL, 5 mmol) were added. The reaction mixture was heated at 60°C for 2 h with continuous stirring and then poured into a beaker containing an ice-water mixture with few drops of HCl. The solid product so formed was filtered off, washed with EtOH and dried. Recrystallization from dioxane afforded compound 6. Yield (70%), mp 274-275 °C (from DMF); IR (KBr) ) cm-1; 3382, 3211 (NH, NH2), 2987, 2845, 2241(CN),1673 (C=O), 1335, 1217 (C=S) ; 1HNMR (DMSO- D2O): δ 4.02 (s, 2H, D2O-exchangeable NH2),7.25–8.48 (m, 10H, ArH), 8.45 (s, 1H, D2O-exchangeable NH); 13C NMR (DMSO-d6) δ:92.4 (C-3-quinoline), 118.9 (CN), 119.1, 119.6, 120.5, 122.2, 123.3, 125.3, 126.1, 126.1, 130.2, 130.2, 127.2, 128.2, 132.7, 144.2, 147.2, 167.6, 168.3(C-2-quinoline)., 193.8(C-2-thiazole), MS (m/z): 403.48 (27.10, M+) Anal. Calcd for C20H13N5OS2: C, 59.54; H, 3.25; N, 17.36, Found: C, 59.28; H, 3.45; N, 17.61.
2.2.6. 3,5-Diamino-4-cyano-N-(3-cyanoquinolin-2-yl)thiophene-2-carboxamide (7)
- To a solution of compound 3 (2.36 g, 5 mmol) in dioxane (25 ml) containing triethylamine (1.00 mL), malononitrile (0.33 g, 5 mmol) was added followed by the addition of an equimolar amount of elemental sulfur (0.16 g, 5 mmol). The reaction mixture was heated under reflux for 5 h, then cooled and neutralized by pouring onto ice/water mixture containing few drops of hydrochloric acid. The solid product formed was collected by filtration and crystallized from dioxane. Yield (80%), mp 256-257°C (from ethanol) ); IR (KBr) ) cm-1; 3495, 3432, 3320 (NH, 2NH2, NH), 2991, 2844, 2250, 2226(2CN), 1647 (C=O); 1HNMR (DMSO- D2O): δ 3.69 (s, 4H, D2O-exchangeable 2NH2), 7.50–8.12(m, 5H, ArH), 9.07 (s, 1H, D2O-exchangeable NH); MS (m/z): 334.36 (16.29, M+) Anal. Calcd for C16H10N6OS: C, 57.48; H, 3.01; N, 25.14, Found: C, 57.70; H, 3.18; N, 25.17.
2.2.7. 2-Cyano-N-(3-cyanoquinolin-2-yl)-2-(5-oxo-3-phenylthiazolidin-2-ylidene) acetamide (8)
- To a stirred solution of potassium hydroxide (0.11 g, 2 mmol) in dimethylformamide (20 ml), compound 3 was added (2.36 g, 2 mmol). After stirring for 30 min, phenylisothiocyanate (0.27 g, 2 mmol) was added to the resulting mixture. Stirring was continued for 6 h. Then chloroacetyl chloride (0.16 mL, 2 mmol) was added. Stirring continued for additional 3 h. Then, the reaction mixture was poured onto ice water. The solid product that formed was filtered off, dried and recrystallized from ethanol to afford compound 8. Yield (65%), mp 290-291 °C(from ethanol) ); IR (KBr)) cm-1;3317 (NH), 2997, 2832, 2208,2241(2CN), 1689,1665 (2C=O); ); 1HNMR (DMSO- D2O): δ 3.19(s, 2H, CH2), 7.29-8.33 (m, 10H, ArH), 10.03 (s, 1H, NH), 13C NMR (DMSO-d6) δ: 69.1(C-2-acetamido), 93.3(C-3-quinoline), 113.5, 113.9, 117.2, 117.6 (CN),119.7, 123.4, 125.1, 126.1, 125.3, 126.1, 129.5, 129.5, 130.2, 130.2, 132.7, 144.8, 147.5, 163.3, 177.8,194(C-5-thiazolidine). MS (m/z): 411.44 (31.12, M+) Anal. Calcd for C22H13N5O2S: C, 64.22; H, 3.18; N, 17.02; Found: C, 64.29; H, 3.32; N, 17.09.
2.2.8. 2-Cyano-N-(3-cyanoquinolin-2-yl)-2-(4-methyl-3-phenylthiazol-2(3H)-ylidene)acetamide (9)
- To a stirred solution of potassium hydroxide (0.11 g, 2 mmol) in dimethylformamide (20 ml) compound 3 was added (2.36 g, 2 mmol). After stirring for 30 min, phenyli-sothiocyanate (0.27 g, 2 mmol) was added to the resulting mixture. Stirring was continued for 6 h, and then bro-moacetone (0.3 mL, 2 mmol) was added. Stirring continued for additional 3 h. Then, the reaction mixture was poured onto ice water. The solid product that formed was filtered off, dried and recrystallized from ethanol to give compound 9. Yield (60%), mp 308-310°C (from DMF); IR (KBr) ) cm-1;3227 (NH), 2991, 2838, 2211, 2238(2CN),1660 (C=O); ); 1HNMR (DMSO- D2O): δ 1.91 (s, 3H, CH3), 6.55 (s, 1H, =CH -thiazole), 7.04-8.13 (m, 10H, Ar-H), 10.23 (s, 1H, NH); MS (m/z): 409.46 (28.20, M+) Anal. Calcd for C23H15N5OS: C, 67.47; H, 3.69; N, 17.10; Found: C, 67.50; H, 3.71; N, 17.16.
2.2.9. 2-Cyano-N-(3-cyanoquinolin-2-yl)-4-oxo-4-phenylbutanamide (10)
- A mixture of compound 3 (2.36 g, 2 mmol, 2 mmol) and phenacyl bromide (0.39 g, 2 mmol) in ethanol (30 ml) containing triethylamine (5 drops) was heated under reflux for 1 h. The formed solid product was filtered off, washed with ethanol, dried and recrystallized from mixture of DMF and ethanol mixture (1:3) to afford compound 10. mp 195-197 °C(from ethanol); IR (KBr) ) cm-1;3350 (NH), 2254, 2224 (2CN), 1680, 1644 (2CO);1HNMR (DMSO) δ 2.51 (d, J = 6.2 Hz, 2H, CH2), 3.12 (t, J = 9.6 Hz, 1H, CH), 7.67-7.97 (m, 10H, Ar-H), 10.18 (s, 1H, NH); MS (m/z): 354.36 (33.18, M+) Anal. Calcd for C21H14N4O2: C, 71.18; H, 3.98; N, 15.81; Found: C, 71.24; H, 3.99; N, 16.01.
2.2.10. 3-Amino-N-(3-cyanoquinolin-2-yl)-6-phenyl-1, 2-dihydropyridazine-4-carboxamide (11)
- A mixture of 10 (3.54 g, 0.01 mol) and hydrazine hydrate 98% (2 mL, 0.02 mmol) was refluxed for 7 h. The reaction mixture was left to cool at room temperature, then was poured onto ice cooled water (100 mL) the resulting solid was filtered off, dried well and recrystallized from ethanol/ DMF mixture (1:3) to give compound 11. mp 229-230°C (from ethanol); IR (KBr)) cm-1;3415, 3321 (NH2), 3325, 3290, 3265 (3NH), 2234 (CN), 1645 (CO);1HNMR (DMSO) δ 5.07 (s, 2H, NH2), 6.57 (s, 1H, =CH), 7.12-8.38 (m, 10H, Ar-H), 9.02 (s, 1H, NH), 10.31 (s, 1H, NHCO), 11.13 (s, 1H, NH); MS (m/z): 368.39 (40.15, M+). Anal. Calcd for C21H16N6O: C, 68.47; H, 4.38; N, 22.81; Found: C, 68.77; H, 4.31; N, 22.55.
2.2.11. 6-Amino-N-(3-cyanoquinolin-2-yl)-3-phenyl-2H-1, 2-oxazine-5-carboxamide (12)
- To a solution of 10 (3.54 g, 0.01 mol) in DMF (20 mL), hydroxylamine hydrochloride (0.7 g, 0.01 mol), sodium carbonate (5 g, 0.02 mol) were added. The reaction mixture was heated under reflux for 4 h and then left to cool. The reaction mixture was poured onto ice cooled water (100 mL) the resulting solid was filtered off, dried well and recrystallized from ethanol/DMF mixture (1:3) to yield compound 12. mp 281-282°C(from dioxane); IR (KBr) ) cm-1;3417, 3352(NH2), 3290, 3265 (2NH) 2251 (CN), 1653 (CO); 1HNMR (DMSO) δ 5.92 (s, 1H, NH2), 6.53 (s, 1H, =CH), 7.01-8.25 (m, 10H, Ar-H), 10.12 (s, 1H, NH-CO), 11.33 (s, 1H, NH), MS (m/z): 369.38 (48.24, M+). Anal. Calcd for C21H15N5O2: C, 68.28; H, 4.09; N, 18.96; Found: C, 68.19; H, 4.20; N, 18.87.
3. Results and Discussion
- The synthetic pathways adopted to obtain the newly synthesized compound 2 that was prepared by nucleophilicsubstitution depending on modification of the reported [29] procedure. Where we aimed to development of efficient protocols for the preparation of biologically active heterocyclic derivatives along with the versatility of the organic synthon [31] we herein report the synthesis of 2-aminoquinoline-3-carbonitrile(2). The strategy and aim of this work depend on preparation of 2 inone-pot procedurevia in situ generation of tetrazolo quinoline, on contrast to other procedure involving separation then thermal decomposition of formed tetrazole [29] in addition, to other procedure involving harsh conditions [32-36] and using a key intermediate 3 for synthesis of some new thiazole, pyrazole, thiophene, oxazine and pyridazine derivatives. We elected to examine the conversion of 1 into 2 with the goals of optimizing reaction conditions, under liquid-liquid phase-transfer conditions using chlorobenzene and water as solvent and tetrabutylammonium bromide as catalyst (Scheme 1), depending on activated aromatic systems such chloro [37a] and a few heteroaromatic systems [37b] can undergo nucleophilic substitution by azide ions. Chloro functionality in 2-position of quinoline-3-carbonitrile moiety was found to be labile towards nucleophilic substitution reactions [29, 31], by addition of sodium azide to this chloro functionality in 2-position it form heterocyclic azide that spontaneously cyclize to give the fused tetrazole form. It well be reported that tetrazole are lipophilic, metabolically stable compounds [38]. In our literature tetrazoles can be synthesized directly by a [3+2] dipolar cycloaddition between an organoazide and C=N of quinoline ring and this reaction occurs through a concerted [39] and regioselective [40] [3+2] cycloaddition. It well be known that phase transfer catalyst technique shows the novelty of using sodium borohydride [41] as an efficient reducing agent for tetrazoles to affords pure product in high yields and offers the advantages of permitting a one-pot conversion of 1 into 2 with very simple operative conditions. Thus, in this modified procedure, synthetic strategies based on phase transfer catalyst and evaluated for the cleavage of tetrazoloquinoline intermediate A (scheme 2) keeping sodium borohydride as a reducing agent [42]. In liquid-liquid phase-transfer conditions, tetrabutylammonium bromide was used as catalyst and chlorobenzene together with water (3:1) was preferred as solvent. Chlorobenzene was used as a solvent to elevate the reaction temperature 200C higher than bromobenzene [28] and this elevation in the temperature of the reaction mixture enhance thermal decomposition of formed tetrazole intermediate and the water increase the rate of this decomposition with unimolecularN2 elimination [42] to produce intermediate B (scheme 2), this elimination was affected by no tetrazole ring substituent are placed [43]. Association of liquid-liquid phase-transfer catalyst with azidolysis by using chlorobenzene and water as solvents proved to be cleaner, rate enhancing and yield improving as compared to direct conversion of chloro to amino functionality under harsh conditions [32-36]. In contrast azidolysis followed by reduction facilitated the indirect amination thus azides and tetrazole followed by reduction can be viewed as latent amino functionalities. The generality of this reaction has been shown by the formation of tetrazole intermediate A (Scheme 2) in good yields according to the reported [29] procedure, but in this literature we aimed to formation of tetrazole in situ that exposed to higher temperature condition where it targeted the use of chlorobenzene because it elevate temperature of the reaction higher than other solvent as bromobenzene. In this stage the elevated temperature cause decomposition of formed tetrazole, then comes the role of water in the reaction where it cause azidolysis of intermediate B (scheme 2) and this accompanied by N2elmination, then comes the role of catalyst where quaternary ammonium halide that dissolved in the aqueous phase undergoes anion exchange with the anion of the reactant dissolved in the aqueous solution. The ion-pair formed can cross the liquid-liquid interface due to its lipophilic nature and diffuses from the interface into the organic phase, this step being the phase-transfer that lead to formation of intermediate D (scheme 2). and the catalyst subsequently, returns to the aqueous phase and the cycle continues. At this important stage, role of sodium borohydride was come as reducing agent to form the intermediate D (scheme2) then reduction was continued, and on complete the reaction time to afford the corresponding 2-aminoquinoline-3-carbonitrile (2) as a key precursor that its IR-spectra of 2 showed the appearance of NH2 Stretching band at 3350 cm-1., which when react with ethylcyano acetate give the key intermediate from which newly synthesized compounds 4-12 were produced. Where IR of 3 revealed the presence of C=O at 1655cm-1 Moreover, the 1HNMR spectrum exhibited singlet at δ 4.28 for the acetamido CH2 and a singlet at δ 6.83 ppm for the amidic NH. By subjecting 3to reaction with diazonium salt the respective hydrazine derivatives 4a-dwere obtained. The appearance of two singlet protons at δ 10.12- 10.42 ppm and another one at δ 11.83-12.88 ppm belong the two NH and both exchangeable with D2O will prove the proposed structures, that cyclized when it react with hydrazine hydrate to afford 5a-dwhich structure were confirmed by the appearance of lower field D2O exchangeable NH2 that prove the cyclization of compound 4 into 5 containingpyrazole ring. When compound 3 react with the phenyl isothiocyanate and elemental sulfur gave the thiazole-2-thione derivative 6, and reaction of cyanoacetamide 3with elemental sulfur and malononitrile gave the thiophene derivative 7 the 1HNMR of the resulting compound 6 was confirmed by appearance of a singlet at δ 4.02 ppm exchangeable with D2O which belong NH2, In addition to another exchangeable one proton at δ 8.45 ppm that belong to the amidic NH, On the other hand the structure of 7 was confirmed from 1HNMR that contain singlet of four protons at δ 3.69 ppm exchangeable with D2O was prove the presence of 2NH2 and presence of another exchangeable proton at δ 9.07 ppm which belong the amidic NH this data will confirm the compound 7. The active methylene group in the cyanoacetamide derivative 3 readily adds to phenyl isothiocyanate in DMF containing potassium hydroxide to give the non-isolable enaminonitrile intermediate potassium salt, which underwent heterocyclization when treated with α-halocarbonyl compounds such as chloroacetyl chloride, or bromoacetone to afford the corresponding thiazole derivatives 8, 9. The assignment of the structures 8, 9 were based on elemental analysis and spectral data, where the IR of 8 showed two stretching band at 1689, 1665cm-1 that confirm the presence of two C=O and two band at 2241, 2208cm-1 due to (2CN) group, In addition 1HNMR spectrum exhibit a singlet of two proton at δ 3.19 ppm belong the methylene proton and one singlet signals at δ 10.03 ppm for NH, but IR spectrum of 9 displayed stretching band at 3227, 2211, 2238 cm-1 belong NH, and two cyano functions, respectively and It’s 1HNMR spectrum displayed new signals at δ 1.91 ppm for methyl protons and a singlet signal at δ 6.55 ppm assignable for (=CH) proton at C-5 of thiazole ring. On treatment of 3 with phenacyl bromide in refluxing ethanol containing a catalytic amount of triethyl amineafforded compound 10 where IR spectrum revealed additional absorption bands for two carbonyl groups appeared at 1680 and 1644 cm-1 and additional absorption bands for two cyano groups appeared at 2254 and 2224 cm-1, while It's 1H NMR spectrum displayed doublet and triplet signals at δ 2.51 and 3.12 ppm for CH2 and CH groups, respectively, the NH proton appeared at δ 10.18 ppm. Compound 10 was utilized as a starting material for further preparation of several hetero rings. Hence, reaction of 10 with hydrazine hydrate or by refluxing with hydroxyl amine hydrochloride afforded 11 and 12 respectively, the structure of 11 was established on the basis of spectroscopic data, where its IR spectrum showed disappearance of absorption to one cyano function, while appearing absorption bands at 3415 and 3321 cm-1 assignable to the formed amino group, three absorptions for three NH groups at 3328, 3293 and 3273 cm-1 also It’s 1H NMR spectrum displayed a singlet broad signal at δ 5.07 ppm assignable to NH2 protons, singlet signal at δ 6.57 ppm for =CH proton and three singlet signals for three NH protons at δ 9.02, 10.31 and 11.13 ppm was established on the basis of spectroscopic data. Also IR spectrum of 12 showed the disappearance of one cyano function while absorption bands at 3417 and 3352 cm-1 assignable to the amino group were appeared beside two absorptions bands for two NH functions at 3290 and 3265 cm-1. It’s 1H NMR spectrum revealed no signals for the adjacent CH2 and CH groups. On the other hand, four singlet signal was appeared at δ 5.92 ppm for NH2 protons, at δ 6.53 ppm for =CH proton, at δ 10.12, δ 11.13 for two NH. The structure of the synthesized product was proven on the basis of their elemental analysis and spectral data. From the previous mentioned discussion it was clear that our synthetic strategies adopted to obtain the newly synthesized compounds 4-12 depending on the regioselective attack on the cyanoacetamido moiety of 3by different reagents, which, in one or two steps added a highly functionalized substituent or heterocyclic ring to the molecule.
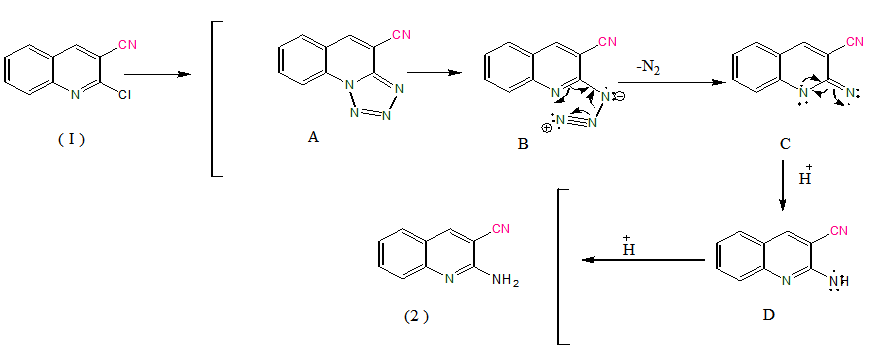 | Scheme 2. Proposde mechanism and possible intermediates |
|
4. Antimicrobial Activity
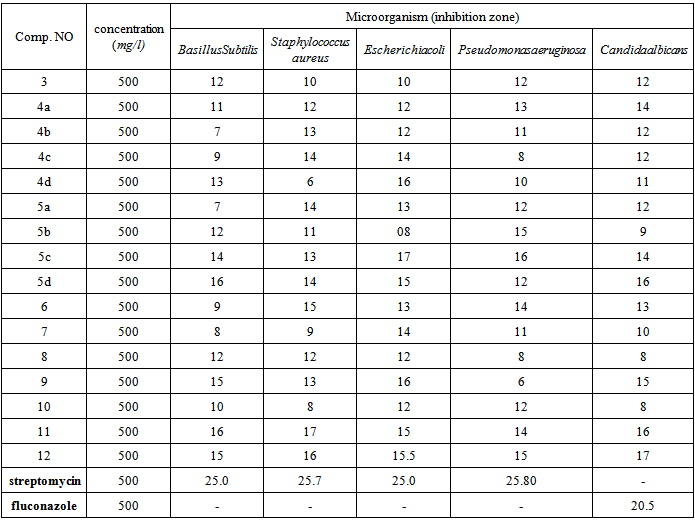
- All newly synthesized compounds were test for their in vitro growth inhibitory activity against a standard strain of two gram positive bacteria viz., Bacillus subtilis, Staphylococci aureus and two gram negative bacteria viz., Escherichia coli, Pseudomonas aeruginosa, in addition to fungi (Candida albicans). Antibacterial activity was done by the disk diffusion method [44]. were the bacteria and fungi sub cultured in BHI medium and incubated for 18h at 37℃, and then the bacterial cells were suspended, according to the McFarland protocol in saline solution to produce a suspended of about 10-5CFU ml 1:10 μ of this suspension was mixed with 10 ml of sterile antibiotic agar at 40℃ and poured onto an agar plate. Five paper disks (6.0mm diameter) were fixed onto nutrient agar plate. The solutions of different compounds under test at a concentration of 500 in 5% DMSO were poured in the cup/well of bacteria seeded agar plates. These plates were incubated at 37oC for 24 hours for E. coli and fungi for 4 days at -2℃, whereas plates of other three bacteria were incubated at 27℃ for 24 hr. The standard antibiotics used were streptomycin (all at 500 μg/ml), and standared antifungal used were fluconazole at 500 μg/ml, The control solution (only 5% DMSO) did not reveals any inhibition. The zone of inhibition produced by each compound was measured in mm. μg/ml and the results of antimicrobials studies are given in Table 1. The discussion and comparison of antibacterial activity were given with respect to streptomycin antibiotic and antifungal screening was compared with fluconazole.
5. Conclusions
- We have synthesized polyfunctionalized heterocyclic systems based on2-cyano-N-(3-cyanoquinolin-2-yl)acetamide (3) using convenient method. The antimicrobial activity of all synthesized compounds showed moderate of antimicrobial activity. From the screening data it was found some derivative 5c, 11 and 12 that containing pyrazole pyridazine and oxazinering have encouraging antifungal activity, which need to be further investigation to get better antifungal and antibacterial agents in future. Microbiological testing of the newel synthesized compounds was performing in the Regional Center for Mycology and Biotechnology, Department of Microbiology, Faculty of Science, Al-Azher University, Cairo, Egypt.