-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2014; 4(1): 1-10
doi:10.5923/j.ajoc.20140401.01
Synthesis and Reactivity of Fluorinated Cyclic Ketones: Initial Findings
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLJoseph C. Sloop1, Matthew Churley1, Alejandro Guzman1, Samantha Moseley1, Stephanie Stalker1, Jonathan Weyand2, Jonathan Yi1
1School of Science and Technology, Georgia Gwinnett College, 1000 University Center Lane, Lawrenceville, GA 30043, USA
2Department of Chemistry and Life Science, United States Military Academy, West Point, NY 10996, USA
Correspondence to: Joseph C. Sloop, School of Science and Technology, Georgia Gwinnett College, 1000 University Center Lane, Lawrenceville, GA 30043, USA.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Using cyclic ketone scaffolds, several novel trifluoroacetylated 1,3-diketones and selectively α-fluorinated ketones have been prepared in yields ranging from 20-77% using the Claisen condensation and electrophilic fluorination by the Selectfluor® reagent, respectively. We report tendencies in the difluorinated and trifluoroacetylated products to form hydrates as well as an unusual range of substrate reactivity toward the Selectfluor® reagent. In general, initial spectroscopic studies suggest that cyclic 2-trifluoroacetylated-1,3-diketones undergo rapid hydration resulting in an equilibrium mixture favoring the diketo hydrate over the keto-enol hydrate. Fluorination of ketone species by Selectfluor® was found to be governed by a combination of steric and electronic effects. Plausible mechanisms for both mono- and difluorination, involving a keto-enol or enolic tautomer, are proposed.
Keywords: 2-trifluoroacetyl-1,3-diketone, Selectfluor®, tautomerism, 19F NMR
Cite this paper: Joseph C. Sloop, Matthew Churley, Alejandro Guzman, Samantha Moseley, Stephanie Stalker, Jonathan Weyand, Jonathan Yi, Synthesis and Reactivity of Fluorinated Cyclic Ketones: Initial Findings, American Journal of Organic Chemistry, Vol. 4 No. 1, 2014, pp. 1-10. doi: 10.5923/j.ajoc.20140401.01.
Article Outline
1. Introduction
- The preparation of molecules with broad appeal in the pharmaceutical, chemical and forensics communities having a ketone moiety is a field of widening interest. For example, certain indanone derivatives are precursors to pharmaceuticals which have anticoagulant properties and industrial products such as near infrared dyes. In addition, the 1,2-indanedione family of compounds are used in the forensic sciences to aid in latent fingerprint elaboration.[1-8]Since fluorine is known to alter bioactivity and other physico-chemical properties in many classes of compounds [6-8], the preparation of new fluorinated examples of these molecules is desirable. Additionally, new insights may be obtained from structure-activity relationship studies and upon determining their bioactivity or efficacy relative to known cases. The preparation and unique keto-enol properties of fluorinated and trifluoromethylated β-diketones have been thoroughly investigated.[9-12] However, the synthesis and study of cyclic 1,3-diketones possessing the trifluoroacetyl group is relatively limited, especially as it relates to the formation of hydrated species.[11-18] Furthermore, while Selectfluor® has found wide application in the preparation of selectively fluorinated molecules[19-21], this work reveals several instances where the usual fluorination conditions fail to produce mono-fluorinated ketone products, lead to more than one fluorinated species or result in difluorination. The desired molecules of interest in this study, which include alicyclic and fused-ring aromatic ketones, are shown in Figure 1. This paper addresses the design and synthetic approach to prepare these fluorinated ketones, interesting hydration properties of these groups as well as the limitations of reactivity and selectivity observed for the Selectfluor® reagent in this series of ketones.
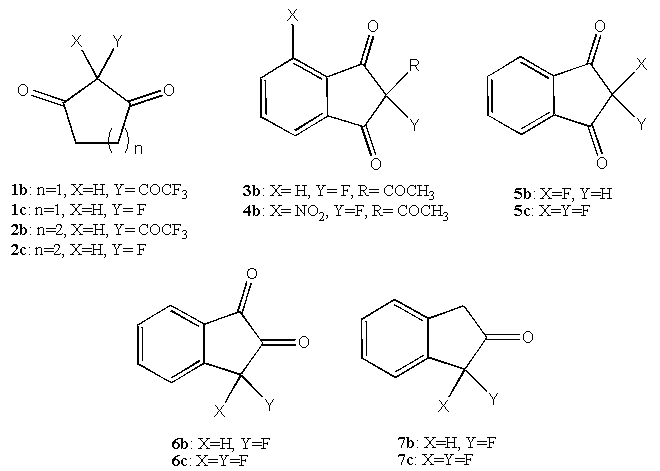 | Figure 1. Target molecules of interest |
2. Results and Discussion
2.1. General Synthesis Results
- Scheme 1 shows the results of our synthesis efforts. Compounds 1a-7a are commercially available and were used without further purification. Compounds 1b and 2b were synthesized in low to moderate yields using a modified Claisen condensation[11,12]. Compounds 1c, 1d, 2c, 2d, 5b, 6c, and 7b were prepared by direct fluorination with Selectfluor® in yields ranging from 10-77%. Attempts to obtain the fluorinated compounds 3b, 4b and 6b were unsuccessful.
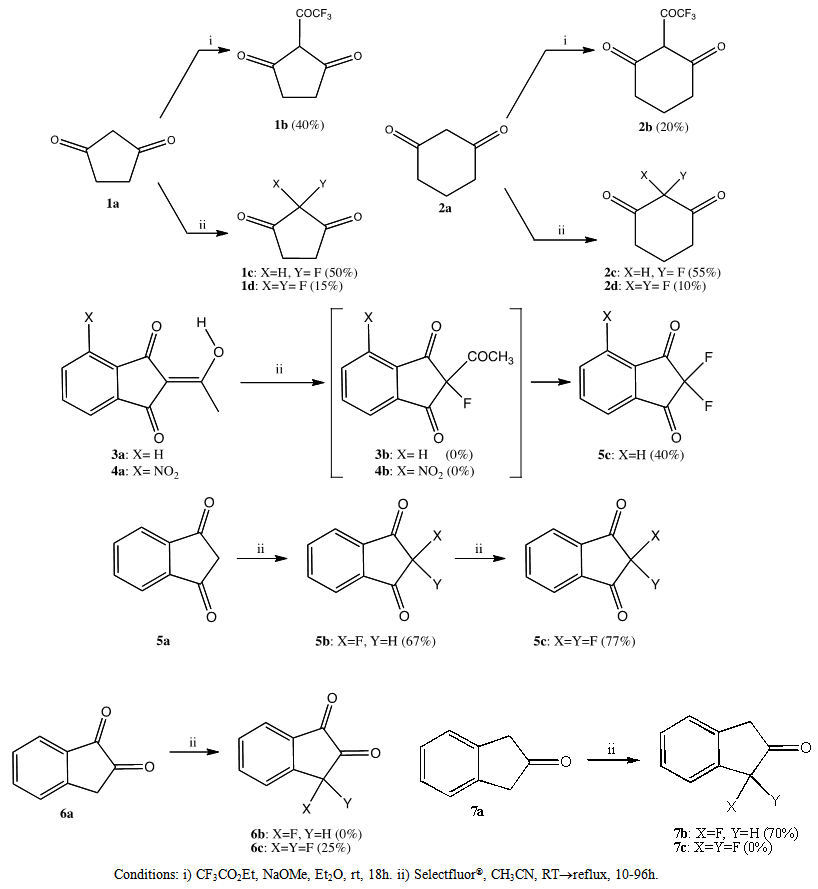 | Scheme 1. Fluorinated ketone synthesis |
2.2. Trifluoracetylation Results
- While the trifluoroacetylation of 1a and 2a provided 1b and 2b in relatively low yields, some interesting results were nonetheless obtained. During the reactions, samples of the reaction mixture were removed at various intervals, neutralized and the reaction progress monitored by 19F NMR. The appearance of fluorine resonances noted ca. δ= -75.7 ppm and -76.3 ppm was consistent with the formation of the keto-enol forms of 1b and 2b, respectively.[22] See Figure 2.
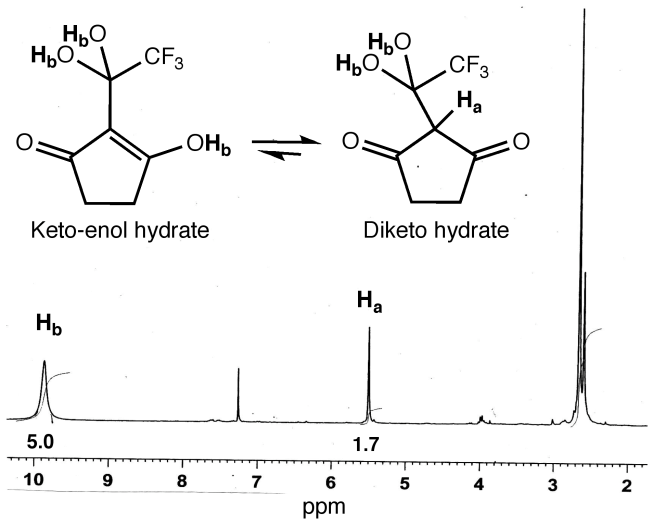 | Figure 2. Proton NMR for 1b hydrate tautomers |
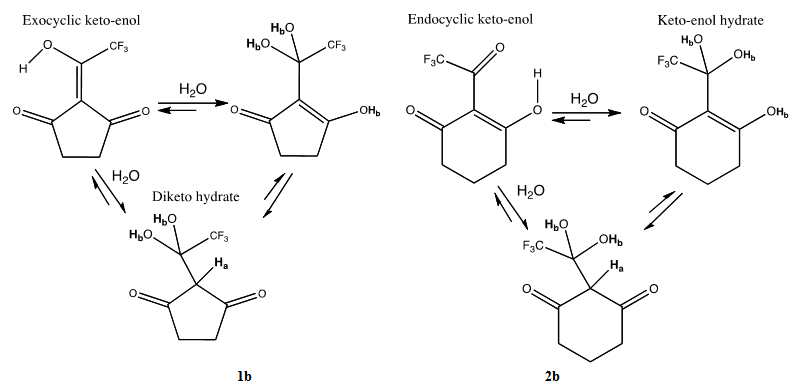 | Scheme 2. Keto-enol hydration product equilibria of 1b and 2b |
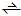 keto-enol hydrate equilibrium. For both 1b and 2b, the diketo hydrate is the dominant form. See Table 1.
keto-enol hydrate equilibrium. For both 1b and 2b, the diketo hydrate is the dominant form. See Table 1.
|
2.3. Fluorination Results and Efficacy Testing
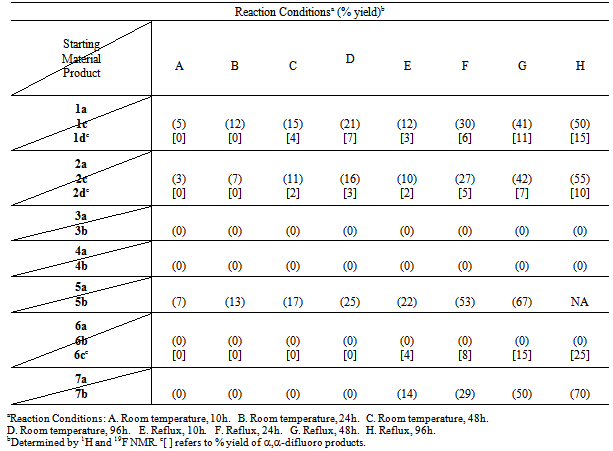
- Standard fluorination conditions employing the Selectfluor® reagent ranged from those conducted at room temperature to reflux with reaction times varying from 10 to 96 h, with acetonitrile serving as the solvent. Reaction progress was monitored by 1H and 19F NMR. To test the efficacy of Selectfluor® as a monofluorination reagent, compounds 1a-7a were subjected to fluorination under a variety of conditions. The results are compiled in Table 2.
|
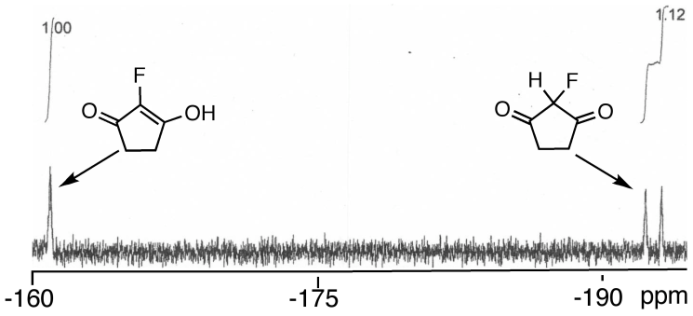 | Figure 3. 19F NMR for 1c tautomers |
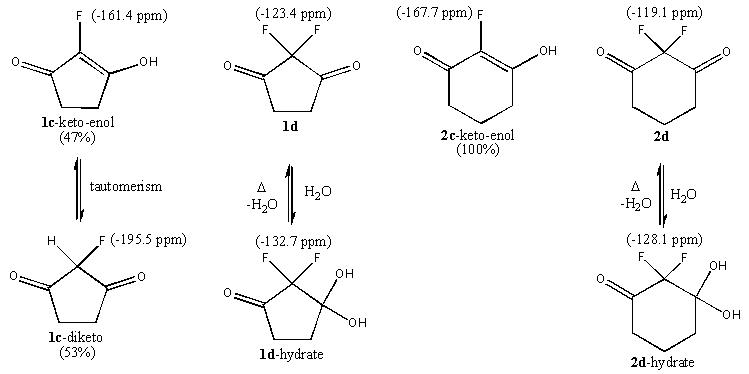 | Figure 4. Fluorination products of 1a and 2a and equilibria exhibited by 1c, 1d, 2c and 2d |
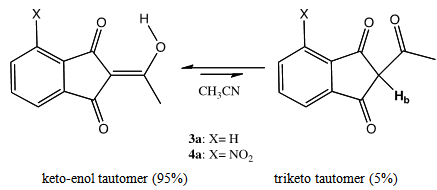 | Figure 5. Keto-enol - triketone tautomeric equilibria of 3a and 4a |
2.4. Plausible Mechanisms for the Electrophilic Fluorination of Cyclic Diketones via Selectfluor®
- The literature indicates considerable disagreement concerning the mechanism by which fluorination occurs with reagents such as Selectfluor®. Polar processes where the “F+” ion is the electrophile and single electron transfer (SET) processes which include a “F. ” radical species have been proposed, but mechanistic studies have not established that a single fluorination mechanism operates in all cases.[20]In the case of the ketone substrates examined in this study, we propose that an enol species initiates the fluorination by having the C=C double bond attack the Selectfluor®. In cases where the enol is formed readily under the reaction conditions or is the major tautomer in the reaction medium, such as cyclic β-diketones 1a and 2a, the fluorination can occur at room temperature, and difluorination can become a complicating issue. In cases where the keto tautomer predominates and/or enolization is slow (e.g. 7a), the reaction is slow even under refluxing conditions. However, multiple fluorinations are not as likely. Finally, fluorination via Selectfluor® does not occur in the usual fashion when the keto-enol substrate is very hindered or very stable (eg. triketones 3a and 4a). Mechanisms for both monofluorination (Scheme 3) and difluorination (Scheme 4) that are consistent with these findings are presented below using compounds 7a and 2b, respectively.
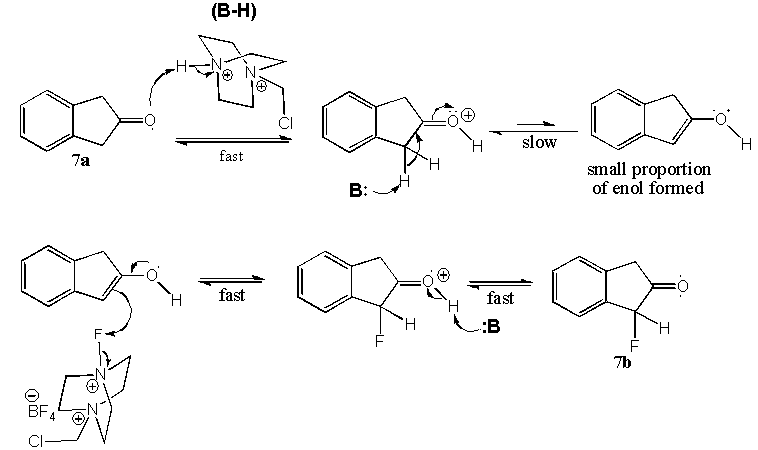 | Scheme 3. Monofluorination mechanism of 7a |
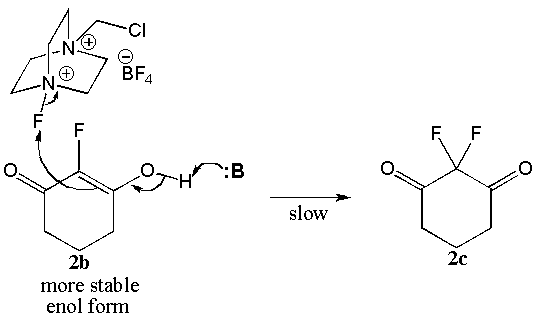 | Scheme 4. Difluorination mechanism of 2b |
 enol tautomeric equilibrium favors the keto form, eg. 5a and 7a. This may also explain the exceptional behavior of 6a, which underwent difluorination even though there was no evidence of enolization during the reaction.
enol tautomeric equilibrium favors the keto form, eg. 5a and 7a. This may also explain the exceptional behavior of 6a, which underwent difluorination even though there was no evidence of enolization during the reaction.3. Conclusions
- This work, while still in its initial stages, provides new insight into several areas of current interest regarding selective trifluoroacetylations and fluorinations of cyclic ketones. Trifluoroacetylations of cyclic β-diketones occur under the usual Claisen condensation conditions, but yields are lower than for their acyclic counterparts. [9,11] Trifluoroacetyl groups are subject to hydration due to the enhanced electrophilicity of the carbonyl and these compounds must be maintained in a moisture-free environment to avoid this phenomenon. Likewise, carbonyl groups adjacent to α,α-difluorinated carbons display a tendency for rapid hydration.Our work has also shed light on the effectiveness of the Selectfluor® reagent in the monofluorination of cyclic ketones, diketones and triketones. Cyclic β-diketones which can tautomerize from the diketo form to a keto-enol form undergo fluorination under mild conditions, but often difluorination can take place as well. When difluorination occurs, hydration at the ketone adjacent to the fluorines frequently takes place. Indanone derivatives such as 3a and 4a are unreactive toward Selectfluor® due to a combination of steric and electronic factors arising from the large size of the fluorinating agent as well as the exceptional stability of the keto-enol tautomer. Conversely, ketones that do not tautomerize appreciably due to instability of the enol form react with the Selectfluor® reagent slowly, if at all, and usually do not give difluorinated products. Notable exceptions to this general rule were the successful fluorinations of 5a and 7a. Likewise, the formation of compound 6b is an apparent departure from this general observation. Further studies are needed to address 6b’s anomalous behavior.Finally, it is possible that either polar or SET fluorination processes may lead to the products in our study. While we were cautious to ensure that reaction vessels were protected from light so as to minimize radical formation, the fact that fluorination was observed in several cases where enolization was not evident could mean that an SET process might be occurring.
4. Experimental
4.1. Chemicals
- All chemicals were obtained from the Aldrich Chemical Company, Eastman Kodak, or Fisher Chemical Company. All solvents and starting materials were checked for purity by mass spectrometry or proton NMR prior to use.
4.2. Instrumentation
- Melting points were obtained on a Mel-Temp melting point apparatus and are uncorrected. NMR data were collected using an Anasazi-90 spectrometer operating at 90.0MHz for 1H, 84.7MHz for 19F and 22.6MHz for 13C or a Brüker Avance 300 spectrometer operating at 300.0MHz for 1H, 288MHz for 19F and 75.4MHz for 13C. Fluorine-19 NMR chemical shifts were referenced to CFCl3 as an external standard. IR data were collected on a Perkin-Elmer FT-IR spectrometer with resolutions of 1cm-1.
4.3. General Procedure for the Preparation of TrifluoroacetylatedDiketones[11,12]
- To 50 mL diethyl ether in a round bottom flask equipped with a magnetic stirrer is added 60 mmol of sodium methoxide slowly. Then, 1eq (60 mmol) of methyl trifluoroacetate is added dropwise slowly while stirring. After 5 minutes, 1eq (60 mmol) of the ketone is added dropwise and stirred overnight at room temperature under a calcium chloride drying tube. The resulting solution is evaporated to dryness under reduced pressure and the solid residue dissolved in 30 mL 3M sulfuric acid. The acidic solution is extracted with 3-15 mL aliquots of diethyl ether, and the organic layer dried over NaSO4. The solvent is evaporated under reduced pressure, the crude diketone purified by distillation, recrystallization or radial chromatography, and purity confirmed by 1H, 13C, and 19F NMR spectroscopy, mass spectrometry and literature melting or boiling points.2-trifluoroacetyl-1,3-cyclopentanedione (1b) This compound was obtained as a 95:5 mixture of exocyclic enol:triketo tautomers as pale yellow crystals (Et2O) in 40% yield, mp 138-140°C. NMR: 1H: δ 2.63 (CH2, 4H, s), 5.41 (keto CH, 1H, s). 13C: δ 30.9, 105.1, 107.2, 115.2 (CF3, q, 1JC-F =274Hz), 159.5 (C-CF3, q, 2JC-F=36Hz), 195.3, 205.0. 19F: δ-75.7. Analysis calcd for C7H5F3O2: C, 43.32, H, 2.60. Found: C, 43.10, H, 2.52. The 1b-hydrate was obtained as a reddish oil (1.7:1 mixture of diketo hydrate:keto-enol hydrate). NMR: 1H: δ2.71 (CH2, 4H, s), 5.58 (CH, 1H, s), 9.88 (OH, 3H, bs). 13C: δ 30.9, 105.1, 107.2, 115.2 (CF3, q, 1JC-F =274Hz), 195.3, 205.0. 19F: δ -82.2 (3F, bs).2-trifluoroacetyl-1,3-cyclohexanedione (2b) This compound was obtained in 20% yield as a beige solid, m.p. 71-74°C. NMR: 1H: δ 1.98-2.6 (CH2, 6H, m). 13C: δ 21.0, 28.8, 30.9, 106.2, 108.1, 117.2 (CF3, q, 1JC-F=271Hz), 162.6 (C-CF3, q, 2JC-F=37Hz), 196.2, 201.0. 19F: δ -76.3 (enol). Analysis calcd for C8H7F3O2: C, 46.16, H, 3.39. Found: C, 46.26, H, 3.31. The 2b-hydrate was obtained as a red-orange oil (1.2:1 mixture of diketo hydrate:keto-enol hydrate.) 1H: δ 1.98 (CH2, 2H, m, J=5.55Hz), 2.35 (CH2, 2H, t, J=6.41Hz), 2.71 (CH2, 2H, t, J=6.59Hz), 5.49 (hydrate, 1H, s), 9.0 (hydrate and enol, 3H, bs). 19F: δ-82.9 (3F, bs).
4.4. General Procedure for the Preparation of Fluorinated Diketones
- A 100mL round bottom flask equipped with a stir bar is charged with 50mL CH3CN and Selectfluor® (0.78 g, 0.0022mol). Once the Selectfluor® has dissolved, the ketone (1eq, 0.0022mol) is added slowly with stirring. The reaction is capped and allowed to run for 10-96 h at room temperature or under reflux at a temperature of 70°C for 10-96 h. The solvent is removed under reduced pressure. The resulting residue is taken up in ~20mL CH2Cl2 and washed with 3 aliquots of ~20 mL distilled water. The organic layers are combined and dried over Na2SO4 and the product purified by radial chromatography or recrystallization.2-fluoro-3-hydroxycyclopent-2-enone and 2-fluoro- 1,3-cyclopentanedione (1c): This compound was obtained as a 52:48 mixture of keto-enol and diketo tautomers in 50% yield as a yellow-brown solid, mp 70-72°C. NMR: 1H: δ 2.36 (t, 3JH-H = 16.2 Hz, 2H), 2.85 (m, 2H), 5.91 (d, 2JH-F = 47.7 Hz, 1H). 13C: δ31.1, 90.8 (d, 1JC-F = 251.3 Hz), 122.3 (d, 1JC-F = 233.9 Hz), 210.1 (d, 2JC-F = 31.0 Hz). 19F: keto-enol: δ-161.4 (s, 1F); diketo: δ-195.5 (d, 2JF-H = 47.7 Hz, 1F). Analysis calcd for C5H5FO2: C, 51.73, H, 4.34. Found: C, 51.48, H, 4.31.2,2-difluoro-1,3-cyclopentanedione (1d): This compound was obtained in 15% yield as tan crystals, mp 41-43°C. NMR: 1H: δ 2.21 (H2C-C=O, 4H, m). 13C: δ 31.1, 110.8 (t, 1JC-F = 248.3 Hz), 199.3 (t, 2JC-F = 28.0 Hz). 19F: δ-123.4 (s, 2F). Analysis calcd for C5H4F2O2: C, 44.79, H, 3.01. Found: C, 44.68, H, 3.12.2,2-difluoro-3,3-dihydroxy-1-cyclopentanone (1d-hydrate): This compound was obtained from the hydration of 1d as beige crystals (ethyl acetate), m.p. 71-74°C [lit. 24] m.p. 70-72°C. NMR: 1H: δ 2.04 (m, 2H), 2.45 (m, 2H), 6.91 (s, hydrate, 2H). 13C: δ 31.0, 32.1, 94.6 (t, 2JC-F = 20.9 Hz), 112.5 (t, 1JC-F = 254.8 Hz), 210.8 (t, 2JC-F = 18.5 Hz). 19F: δ -132.7 (s, 2F).2-fluoro-3-hydroxycyclohex-2-enone (2c): This compound was obtained in 55% yield as pale tan crystals (ethyl acetate), m.p. 150-152°C [lit. 25] (m.p. 152-153°C). NMR: 1H: δ 1.90-2.50 (m, 6H), 11.4 (bs, 1H). 13C: δ20.1, 22.2, 33.0, 106.1, 37.9, 139.8.1 (d, 1JC-F = 235.3 Hz), 200.1 (d, 2JC-F = 14.5 Hz). 19F: δ-167.7 (s, 1F). 2,2-difluoro-1,3-cyclohexanedione (2d): This compound was obtained in 10% yield as pale tan crystals (ethyl acetate), m.p. 68-70°C [lit. 25] (m.p. 70°C). NMR: 1H: δ 1.9 (m, 2H), 2.4 (m, 4H). 13C: δ 18.0, 37.9, 114.1 (t, 1JC-F = 257.6 Hz), 195.2 (t, 2JC-F = 23.5 Hz). 19F: δ-119.0 (s, 2F).2,2-difluoro-3,3-dihydroxy-1-cyclohexanone (2d-hydrate): This compound was obtained from the hydration of 2d as pale tan crystals (ethyl acetate), m.p. 68-76°C [lit. 25] NMR: 1H: δ 1.85 (m, 2H), 2.11 (m, 2H), 2.49 (m, 4H). 13C: δ 20.0, 35.0, 96.0 (t, 1JC-F = 257.6 Hz), 114.2 (t, 1JC-F = 257.0 Hz), 197.3 (t, 2JC-F = 24.5 Hz). 19F: δ -128.1 (s, 2F).2-fluoro-1,3-indanedione (5b). This compound was obtained in 67% yield as pale yellow crystals (EtOH), m.p. 95–97°C [lit 19] (m.p. 96–98°C). NMR: 1H: δ 5.4 (d, 2JH-F = 51.0 Hz, 1H), 7.65–8.22 (m, 4H). 13C: δ 90.1 (d, 1JC-F = 211.2 Hz), 125.3, 138.9, 141.9, 193.5 (d, 2JC-F = 24.0 Hz). 19F: δ −207.3 (d, 2JF-H = 51.1 Hz, 1F).2,2-difluoro-1,3-indanedione (5c). This compound was obtained in 77% yield as a yellowish-brown solid (EtOH), m.p. 115–117°C [lit 21] (m.p. 117–118°C). NMR: 1H: δ 8.0–8.18 (m, 4H). 13C: δ 104.1 (t, 1JC-F = 264 Hz), 128.6, 138.1, 139.2 (t, 3JC-F = 4.3 Hz), 185.8 (t, 2JC-F = 24.1Hz). 19F: δ −125.8 (s, 2F).3,3-difluoro-1,2-indanedione (6b): This compound was obtained in 25% yield as yellow-brown crystals, m.p. 99-103°C, dec. NMR: 1H: δ 7.1-8.6 (m, 4H). 19F: δ-86.1 (s, 2F). Purification by column chromatography resulted in decomposition of the product, so further characterization was not possible at the time of this publication.(±)1-fluoro-2-indanone (7b) This compound was obtained in 70% yield as a racemic mixture of enantiomers as a brown oil [lit 29]. NMR: 1H: δ 3.58 (s, 2H), 5.7 (d, 2JH-F =52.5Hz, 1H), 7.0-7.6 (m, 4H). 19F: δ-180.8 (d, 2JF-H =52.5Hz, 1F).
ACKNOWLEDGEMENTS
- The authors thank Georgia Gwinnett College SS&T STEC 4500 Research Seed Grant program and the United States Army Research Laboratory (C&LS-2) for the funding of this work.