-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2013; 3(1): 16-23
doi:10.5923/j.ajoc.20130301.03
Utility of 6-Aryl-5-Cyano-2-Thiouracil Derivative as a Precursor for the Synthesis of Some New Pyrimidines
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLEssam Abdelghani, Said Aly Said, M. G. Assy, Atef M. Abdel Hamid
Chemistry Department, Faculty of Science, Zagazig University, Zagazig, Egypt
Correspondence to: Atef M. Abdel Hamid, Chemistry Department, Faculty of Science, Zagazig University, Zagazig, Egypt.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Reactions of 6-aryl-5-cyano-2-thiouracil derivatives 1a,b with a variety of reagents leads to the synthesis of some new pyrimidines and condensed pyrimidines. The antibacterial activity was studied against examples of Gram-positive and Gram-negative bacteria.
Keywords: Tetrazolopyrimidine, Diaminopyrimidine, Triazolopyrimidine, Thienopyrimidine, Antibacterial
Cite this paper: Essam Abdelghani, Said Aly Said, M. G. Assy, Atef M. Abdel Hamid, Utility of 6-Aryl-5-Cyano-2-Thiouracil Derivative as a Precursor for the Synthesis of Some New Pyrimidines, American Journal of Organic Chemistry, Vol. 3 No. 1, 2013, pp. 16-23. doi: 10.5923/j.ajoc.20130301.03.
Article Outline
1. Introduction
- It is well known that pyrimidine derivatives are of great biological interest, especially as antimicrobial[1-4], Anti-HIV[5, 6], anti-inflammatory[7, 8] and antitumor agents[9-18]. Fused pyrimidines continue to attract considerable attention because of their great practical usefulness, primarily due to very wide spectrum of biological activities. This is evident in particular from publications of regular reviews on the chemistry of systems where the pyrimidine ring is fused to various heterocycles such as purines, pteridines, quinazolines, pyridopyrimidines, triazolo pyrimidines, pyrazolopyrimidines, pyrimidoazepines, furopyrimidines and pyrolopyrimidines. Many simple fused pyrimidines such as purines and pteridines are biologically active by themselves[19, 20], or are essential components of very important naturally occurring substances (i.e., nucleic acids). Some pteridine derivatives are also used as anti-leukemic drugs[21], or potassium- conserving diuretics[22]. In addition, several quinazoline alkaloids exhibit hypnotic[23, 24], bronchodilatory[25], and antimalarial[26, 27] activity. 2-Thiouracils and6-aryl-2-thiouracils are well known for their antimicrobial, anticancer and antiviral activity[28-30]. The aim of this study was to synthesize some new pyrimidine derivatives based on 6-aryl-5-cyano-2-thiouracil derivative and to investigate their antibacterial activity.
2. Results and Discussion
2.1. Chemistry
- In connection with our programme aiming to the synthesis and evaluate the biological activity of fused heterocycles [31-33], we have tried to prepare some condensed pyrimidines. Thioxopyrimidine 1 was condensed with chloroacetic acid and p-chlorobenzaldehyde in a mixture of acetic acid and acetic anhydride in the presence of fused sodium acetate to yield thiazolopyrimidine derivative 2[34]. The structure of compound 2 was confirmed by its analytical and spectral data. IR spectrum of 3 showed absorption bands at 2219, 1761 and 1694 cm-1 corresponding to C≡N and C=O groups, respectively. Its 1H NMR spectrum showed signals at δ = 7.65-8.22 ppm corresponding to Ar-H and ethylenic proton.The alkylation of methylthioxopyrimidine derivative 1b using chloroacetonitrile gave N-alkylated derivative 3. Its IR spectrum showed absorption bands at 2224 and 1686 cm-1 corresponding to C≡N and C=O groups, respectively. While its 1H NMR spectrum showed two singlets at δ = 2.77 and 5.20 ppm corresponding to SCH3 and CH2, respectively.Hydrazinolysis of 3 with hydrazine hydrate gave diaminopyrimidine derivative 4 via rearrangement[35]. IR spectrum of 4 showed absorption bands at 3428, 3330 cm-1 corresponding to NH and NH2, in addition to two bands at 2209 and 1651 cm-1 corresponding to C≡N and C=O groups, respectively. Its 1H NMR spectrum showed three singlets at δ = 4.15, 4.41 and 11.50 ppm for NH2, CH2 and NH, respectively.The cyclocondensation of 4 with formic acid afforded triazolopyrimidine derivative 5[36]. The structure of 5 was elucidated by its IR and 1H NMR spectra. IR spectrum showed absorption bands at 2221 and 1698 cm-1 corresponding to C≡N and C=O groups, respectively. Whereas 1H NMR spectrum showed two singlets at δ = 5.14 and 8.8 ppm corresponding to CH2 and N=CH, respectively. Treatment of compound 4 with nitrous acid at 0 oC gave tetrazolopyrimidine derivative 6. IR spectrum of compound 6 showed absorption bands at 2229 and 1707 cm-1 corresponding to C≡N and C=O, respectively. Its 1H NMR spectrum showed one singlet at δ = 5.33 ppm corresponding to CH2 (Scheme 1).Chlorination of pyrimidone 1b using phosphorous oxychloride produced chloropyrimidine derivative 7[37] in which Position 4 showed distinct activity and the chlorine atom could be replaced by aromatic amines namely, anthranilic acid, p-aminoacetophenone and/or o-phenylenediamine to give 4-aminoaryl pyrimidine derivatives 8a-c[38], respectively. The structures of compounds 8a-c were confirmed by their analytical and spectral data. IR spectrum of compound 8a showed absorption bands at 3438, 2210 and 1674 cm-1 corresponding to OH, C≡N and C=O groups, respectively. Its 1H NMR spectrum showed three singlets at δ = 2.60, 11.89 and 13.92 ppm corresponding to SCH3, NH and COOH, respectively. IR spectrum of compound 8b showed absorption bands at 2209 and 1676 cm-1 corresponding to C≡N and C=O groups, respectively. Its 1H NMR spectrum showed three singlets at δ = 2.50, 2.56 and 10.11 ppm corresponding to SCH3, COCH3 and NH, respectively. IR spectrum of compound 8c showed absorption bands at 3422, 3308 cm-1 corresponding to NH and NH2 groups, in addition to a band at 2209 cm-1 corresponding to C≡N group. Its Mass spectrum showed M+ at 367 (40%).Compound 8a underwent intramolecular cyclocondensation, when heated with acetic anhydride to give pyrimidoqinazoline derivative 9[39]. IR spectrum showed absorption bands at 2217 and 1693 cm-1 corresponding to C≡N and C=O groups, respectively. Its 1H NMR spectrum showed one singlet at δ = 2.58 ppm corresponding to SCH3. Treatment of com-pound 8c with nitrous acid at 0℃ yielded benzotriazolylpyrimidine derivative 10. IR spectrum of compound 10 showed an absorption band at 2215 cm-1 corresponding to C≡N group. Its 1H NMR spectrum showed one singlet at δ = 2.77 ppm corresponding to SCH3. Reaction of 7 with hydrazine hydrate afforded dihydrazinylpyrimidine derivative 11[39]. The structure of 11 was elucidated by: (a) IR spectrum which showed absorption bands at 3400, 3303 cm-1 corresponding to NH and NH2 groups, in addition to one absorption band at 2211 cm-1 corre-sponding to C≡N group. Its 1H NMR spectrum showed two singlets at δ = 4.50 and 4.72 ppm corresponding to two NH2, in addition to two singlets at δ = 8.85 and 11.87 ppm corresponding to two NH.
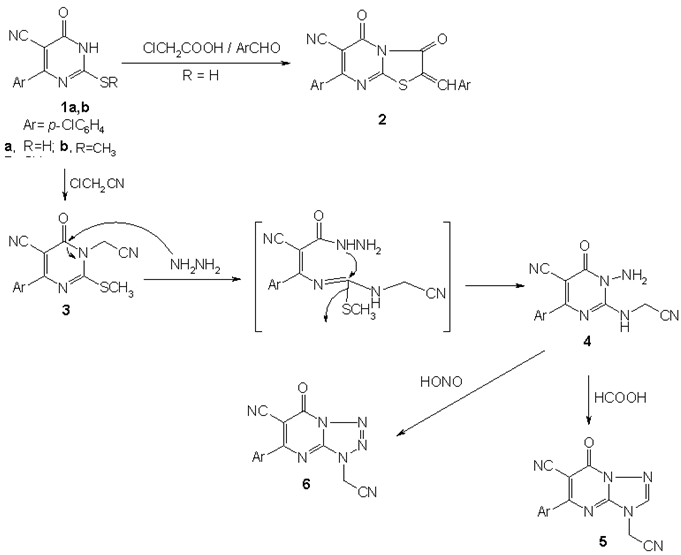 | Scheme 1. Heterocyclization of thiouracil derivatives to azolopyrimidines 2,5 and 7 |
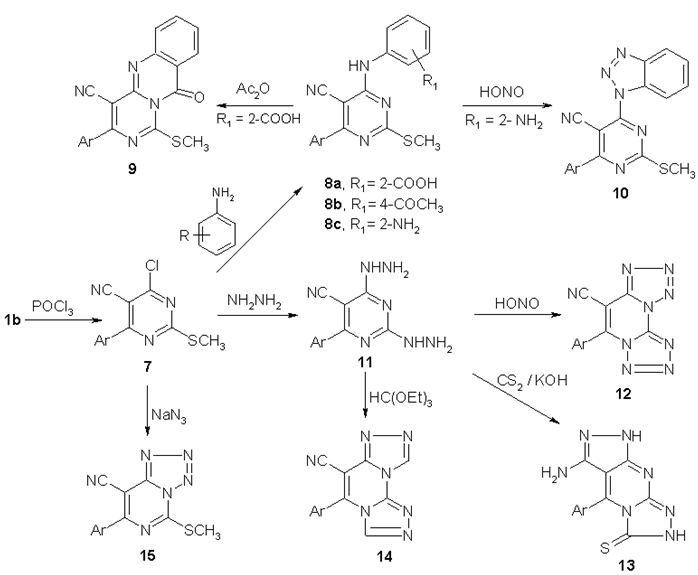 | Scheme 2. Heterocyclization of compound 7and 11 |
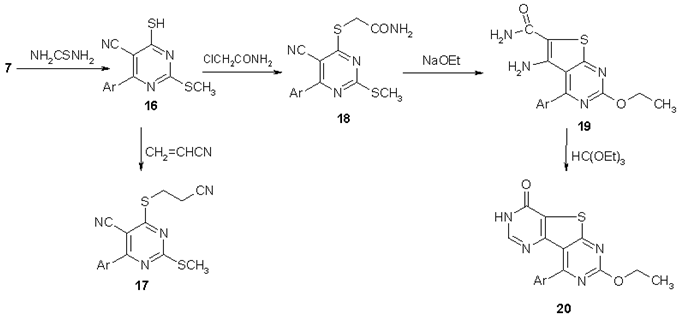 | Scheme 3. Heterocyclization of compound 16 |
2.2. Antibacterial Activity
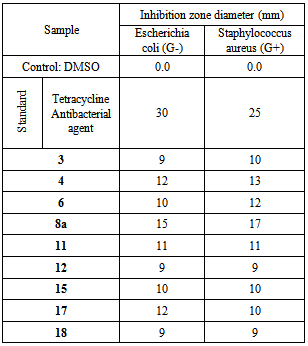
- Compounds 3, 4, 6, 8a, 11, 12, 15, 17 and 18 were tested for in vitro an-timicrobial activity. Tetracycline was used as antibacterial agent ent standard. The zone of inhibition of bacterial growth around the disc was observed. The screening results given in Table 1 indicate that all the tested compounds have antibacterial activities against Escherichia coli and Staphylococcus aureus.
|
3. Experimental
- All melting points are uncorrected. IR spectra (KBr) were run on a Unicam SP 1200G infrared spectrophotometer. 1H NMR spectra (DMSO-d6) were run on a Varian spectrometer (300 MHz) with a T.M.S. as internal standard. Elemental analyses and in vitro antimicrobial activities were carried out at Micro Analytical Center, Cairo University. Compounds 1a,b, 7 and 16 were prepared by the procedures described in literature[37, 45].7-(4-chlorophenyl)-2-[(4-chlorophenyl)methylidene]-3,5-dioxo-2,3-dihydro-5H-[1,3]thiazolo[3,2-a]pyrimidine-6-carbonitrile (2). A mixture of 1a (0.01 mol), chloroacetic acid (0.01 mol), p-chloro benzaldehyde (0.01 mol) and fused sodium acetate (2g) in glacial acetic acid (30 ml) and acetic anhydride (15 ml) was refluxed for 4h, cooled and The solid formed was filtered off, dried and recrystallized from acetic acid to give 2 as yellow crystals. m.p. > 300oC, Yield: 74% . IR (KBr) υmax: 3086 (CH), 2219 (C≡N), 1761, 1694 (C=O) cm-1. 1H NMR (DMSO-d6) δ: 7.65-8.22 (m, 9H, Ar-H + ethylenic proton) ppm. Anal. Calcd. for C20H9Cl2N3O2S (426.28): C, 56.35; H, 2.13; N, 9.86. Found: C, 56.28; H, 2.21; N, 9.77.4-(4-Chlorophenyl)-1-(cyanomethyl)-2-(methylthio)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (3)A mixture of 1b (0.01 mol), chloroacetonitrile (0.01 mol) and K2CO3 (0.01 mol) in DMF (30 ml) was heated on a water bath for 5h, cooled and poured onto cold water. The solid formed was filtered off, washed with water then dried and recrystallized from EtOH to give 3 as brown crystals. m.p. 192-194 oC, Yield: 63%. IR (KBr) υmax: 3085, 2951(CH), 2224 (C≡N), 1686 (C=O) cm-1. 1H NMR (DMSO-d6) δ: 2.77 (s, 3H, CH3), 5.20 (s, 2H, CH2), 7.67-8.06 (m, 4H, Ar-H) ppm. Anal. Calcd. for C14H9ClN4OS (316.76): C, 53.08; H, 2.86; N, 17.69. Found: C, 53.13; H, 2.81; N, 17.62.1-Amino-4-(4-chlorophenyl)-2-((cyanomethyl)amino)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (4)A mixture of 3 (0.01 mol) and hydrazine hydrate (3 ml) in EtOH (50 ml) was refluxed for 4h, cooled and the solid formed was filtered off, dried and recrystallized from EtOH to give 4 as yellow crystals. m.p. > 300 oC, Yield: 73%. IR (KBr) υmax: 3428, 3330 (NH, NH2), 3070 (CH) and 2209 (C≡N), 1651 (C=O) cm-1. 1H NMR (DMSO-d6) δ: 4.15 (s, br., 2H, NH2), 4.41 (s, 2H, CH2), 7.59-7.96 (m, 4H, Ar-H), 11.50 (s, br., 1H, NH) ppm. Anal. Calcd. for C13H9ClN6O (300.70): C, 51.92; H, 3.02; N, 27.95. Found: C, 52.01; H, 2.97; N, 27.89.5-(4-chlorophenyl)-3-(cyanomethyl)-7-oxo-3,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine-6-carbonitrile (5)A mixture of 4 (0.01 mol) and formic acid (20 ml) was refluxed for 6h. The formed precipitate after cooling was filtered off, dried and recrystallized from EtOH to give 5 as brown crystals. m.p. > 300 oC, Yield: 67% . IR (KBr) υmax: 3104 (CH), 2221 (C≡N), 1698 (C=O), 1643 (C=N), 1601 (C=C) cm-1. 1H NMR (DMSO-d6) δ: 5.14 (s, 2H, CH2), 7.67-7.75 (m, 4H, Ar-H), 8.80 (N=CH) ppm. Anal. Calcd. for C14H7ClN6O (310.70): C, 54.12; H, 2.27; N, 27.05. Found: C, 54.17; H, 2.21; N, 26.98.5-(4-chlorophenyl)-3-(cyanomethyl)-7-oxo-3,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carbonitrile (6)A stirred ice cold solution of compound 4 (0.01 mol) in acetic acid (20 ml) was treated drop wise with a cold solution of NaNO2 (0.01 mol) in water (5 ml). The reaction mixture was further stirred for 30 min. then poured into cold water and the separated solid product was filtered off, washed with water, dried and recrystallized from EtOH to give 6 as brown crystals. m.p. 246-248 oC, Yield: 85%. IR (KBr) υmax: 3086 (CH), 2229 (C≡N), 1707 (C=O), 1614 (C=N) cm-1. 1H NMR (DMSO-d6) δ: 5.33 (s, 2H, CH2) 7.68-8.00 (m, 4H, Ar-H) ppm. Anal. Calcd. for C13H6ClN7O (311.69): C, 50.09; H, 1.94; N, 31.46. Found: C, 50.17; H, 1.91; N, 31.52.2-{[6-(4-chlorophenyl)-5-cyano-2-(methylsulfanyl)pyrimidin-4-yl]amino}benzoic acid (8a)A mixture of 7 (0.01 mol) and anthranilic acid (0.01 mol) in acetic acid (30 ml) was refluxed for 10h and the solid formed on hot was filtered off, dried and recrystallized from n-butanol to give 8a as white crystals. m.p. 268-270 oC, Yield: 77%. IR (KBr) υmax: 3438 (OH), 3115 (NH), 2929 (CH), 2210 (C≡N), 1674 (C=O), 1621(C=N) cm-1. 1H NMR (DMSO-d6) δ: 2.60 (s, 3H, CH3), 7.21-8.71(m, 8H, Ar-H), 11.89(s, 1H, NH), 13.92(s, 1H, COOH) ppm. Anal. Calcd. for C19H13ClN4O2S (396.85): C, 57.50; H, 3.30; N, 14.12. Found: C, 57.56; H, 3.25; N, 14.17.4-((4-acetylphenyl)amino)-6-(4-chlorophenyl)-2-(methylthio)pyrimidine-5-carbonitrile (8b) and 4-((2-aminophenyl)amino)-6-(4-chlorophenyl)-2-(methylthio)pyrimidine-5-carbonitrile (8c). A mixture of 7 (0.01mol),p-aminoacetophenone and/or o-phenylenediamine (0.01 mol) and K2CO3 (0.01 mol) in EtOH (50 ml) was refluxed for 3h and The solid formed on hot was filtered off, dried and recrystallized from the proper solvent to give 8b and 8c, respectively.Compound 8bFrom n-butanol as yellow crystals. m.p. 256-258 oC, Yield: 65%. IR (KBr) υmax: 3305 (NH), 2209 (C≡N), 1676 (C=O), 1604 (C=N) cm-1. 1H NMR (DMSO-d6) δ: 2.50 (s, 3H, SCH3), 2.56 (s, 3H, COCH3), 7.64-7.98 (m, 8H, Ar-H), 10.11 (s, br., 1H, NH) ppm. Anal. Calcd. for C20H15ClN4OS (394.88): C, 60.83; H, 3.83; N, 14.19. Found: C, 60.87; H, 3.79; N, 14.23.Compound 8cFrom n-butanol as yellow crystals. m.p. 220-222 oC, Yield: 86%. IR (KBr) υmax: 3422, 3308 (NH2, NH), 2209 (C≡N), 1624 (C=N) cm-1. MS: m/z = 367 (M+), 352 (M+-CH3), 351 (M+-NH2), 332 (M+-Cl), 320 (M+-SCH3). Anal. Calcd. for C18H14ClN5S (367.86): C, 58.77; H, 3.84; N, 19.04. Found: C, 58.82; H, 3.78; N, 18.96.3-(4-chlorophenyl)-1-(methylthio)-10-oxo-10H-pyrimido[6,1-b]quinazoline-4-carbonitrile (9). A mixture of 8a (0.01 mol) and acetic anhydride (30 ml) was refluxed for 6h then cooled and the precipitate formed was filtered off, dried and recrystallized from acetic acid to give 9 as yellow crystals. m.p. 262-264oC, Yield: 48%. IR (KBr) υmax: 3073 (CH), 2217 (C≡N), 1693 (C=O), cm-1. 1H NMR (DMSO-d6) δ: 2.58 (s, 3H, SCH3), 7.53-8.22 (m, 8H, Ar-H) ppm. Anal. Calcd. for C19H11ClN4OS (378.83): C, 60.24; H, 2.93; N, 14.79. Found: C, 60.30; H, 2.89; N, 14.824-(1H-benzotriazol-1-yl)-6-(4-chlorophenyl)-2-(methylthio)pyrimidine-5-carbonitrile (10). A stirred cold solution of compound 8c (0.01 mol) in conc. HCl (20 ml) was treated drop wise with a cold solution of NaNO2 (0.01 mol) in water (5 ml). The reaction mixture was further stirred for 30 min. then poured into cold water and the separated solid product was filtered off, washed with water, dried and recrystallized from n-butanol to give 10 as white crystals. m.p. 244-246 oC, Yield: 63%. IR (KBr) υmax: 3079 (CH), 2215 (C≡N) cm-1. 1H NMR (DMSO-d6) δ: 2.77 (s, 3H, SCH3), 7.67-8.42 (m, 8H, Ar-H) ppm. Anal. Calcd. for C18H11ClN6S (378.84): C, 57.07; H, 2.93; N, 22.18. Found: C, 57.12; H, 2.89; N, 22.15.4-(4-chlorophenyl)-2,6-bis(hydrazino)pyrimidine-5-carbonitrile (11). A mixture of 7 (0.01 mol) and hydrazine hydrate (5 ml) in ethanol (50 ml) was refluxed for 6h. The formed precipitate after cooling was filtered off, dried and recrystallized from n-butanol to give 11 as yellow crystals. m.p. 240-242 oC, Yield: 86%. IR (KBr) υmax: 3400, 3303, (NH, NH2), 3099, 3054 (CH), 2211 (C≡N) cm-1. 1H NMR (DMSO-d6) δ: 50 (s, br., 2H, NH2), 4.72 (s, br., 2H, NH2), 7.55-7.85 (m, 4H, Ar-H), 8.85 (s, br., 1H, NH), 11.87 (s, br., 1H, NH) ppm. Anal. Calcd. for C11H10ClN7 (275.70): C, 47.92; H, 3.66; N, 35.56. Found: C, 47.88; H, 3.71; N, 35.62.5-(4-chlorophenyl)bistetrazolo[1,5-a:1',5'-c]pyrimidine-6-carbonitrile (12). A stirred cold solution of compound 11 (0.01 mol) in acetic acid (50 ml) was treated drop wise with a cold solution of NaNO2 (0.01 mol) in water (5 ml). The reaction mixture was further stirred for 30 min. then poured into cold water and the separated solid product was filtered off, washed with water, dried and recrystallized from EtOH to give 12 as white crystals. m.p. 144-146 oC, Yield: 73%. IR (KBr) υmax: 3084 (CH), 2222 (C≡N) cm-1. 1H NMR (DMSO-d6) δ: 7.69-8.01 (m, 4H, Ar-H) ppm. Anal. Calcd. for C11H4ClN9 (297.66): C, 44.39; H, 1.35; N, 42.35. Found: C, 44.42; H, 1.31; N, 42.39.6-amino-5-(4-chlorophenyl)-2,8-dihydro-3H-pyrazolo[3,4-d][1,2,4]triazolo[4,3-a]pyrimidine-3-thione (13). A mixture of 11 (0.01 mol), CS2 (5 ml) and KOH (0.02 mol) in ethanol (75 ml) was refluxed for 6h. After removal of the solvent under vacuum, water was added and the alkaline solution was neutralized with diluted HCl. The formed precipitate was filtered off, washed with water then dried and recrystallized from DMF/EtOH to give 13 as yellow crystals. m.p. > 300 oC, Yield: 62%. IR (KBr) υmax: 3434, 3300 (NH, NH2), 1387 (C=S) cm-1. 1H NMR (DMSO-d6) δ: 6.02 (s, br., 2H, NH2), 7.61-7.95 (m, 4H, Ar-H), 12.27 (s, br., 1H, NH), 13.96 (s, br., 1H, NH) ppm. Anal. Calcd. for C12H8ClN7S (317.76): C, 45.36; H, 2.54; N, 30.86. Found: C, 45.42; H, 2.49; N, 30.91.5-(4-chlorophenyl)bis[1,2,4]triazolo[4,3-a:4',3'-c]pyrimidine-6-carbonitrile (14). A mixture of 11 (0.01 mol) and triethyl orthoformate (10 ml) in acetic acid (30 ml) was refluxed for 4h. The precipitate formed during heating was filtered off, dried and recrystallized from DMF to give 14 as orange crystals. m.p. > 300 oC, Yield: 54%. IR (KBr) υmax: 3076 (CH), 2237(C≡N), 1630(C=C), 1589 (C=N) cm-1. 1H NMR (DMSO-d6) δ: 7.80-7.87 (m, 4H, Ar-H), 9.10 (s, 1H, N=CH), 10.06 (s, 1H, N=CH) ppm. Anal. Calcd. for C13H6ClN7 (295.69): C, 52.81; H, 2.05; N, 33.16. Found: C, 52.78; H, 2.10; N, 33.12.7-(4-chlorophenyl)-5-(methylthio)tetrazolo[1,5-c]pyrimidine-8-carbonitrile (15). A mixture of 7 (0.01 mol) and sodium azide (0.01 mol) in acetic acid (30 ml) was refluxed for 4h, cooled and the formed precipitate was filtered off, dried and recrystallized from EtOH to give 15 as white crystals. m.p. 294-296 oC,Yield:73%. IR (KBr) υmax: 2921 (CH), 2201(C≡N) cm-1. 1H NMR (DMSO-d6) δ: 2.59 (s, 3H, SCH3), 7.62-7.99 (m, 4H, Ar-H) ppm. Anal. Calcd. for C12H7ClN6S (302.74): C, 47.61; H, 2.33; N, 27.76; Found: C, 47.58; H, 2.37; N, 27.81.4-(4-chlorophenyl)-6-[(2-cyanoethyl)thio]-2-(methylthio)pyrimidine-5-carbonitrile (17). A mixture of 16 (0.01 mol), acrylonitrile (0.01 mol) and few drops of piperidine in n-butanol (30 ml) was refluxed for 3h, cooled and the formed precipitate was filtered off, dried and recrystallized from n-butanol to give 17 as white crystals. m.p. 152-154 oC, Yield: 78%. IR (KBr) υmax: 3076, 2993 (CH), 2211(C≡N) cm-1. 1H NMR (DMSO-d6) δ: 2.64 (s, 3H, SCH3), 3.04 (t, 2H, CH2), 3.61 (t, 2H, CH2), 7.65-7.96 (m, 4H, Ar-H) ppm. Anal. Calcd. for C15H11ClN4S2 (346.86): C, 51.94; H, 3.20; N, 16.15. Found: C, 51.89; H, 3.23; N, 16.18.2-{[6-(4-chlorophenyl)-5-cyano-2-(methylthio)pyrimidin-4-yl]thio}acetamide (18). A mixture of 16 (0.01 mol), cyanoacetamide (0.01 mol) and anhydrous sodium acetate (0.01 mol) in absolute ethanol (50 ml) was refluxed for 3h, cooled and the formed precipitate was filtered off, dried and recrystallized from n-butanol to give 18 as white crystals. m.p. 220-222 oC, Yield: 77%. IR (KBr) υmax: 3370, 3188 (NH2), 2978 (CH), 2209 (C≡N), 1660 (C=O), 1597 (C=N) cm-1. 1H NMR (DMSO-d6) δ: 2.61 (s, 3H, SCH3), 4.06 (s, 2H, SCH2), 7.25 (s, br., 2H, CONH2), 7.65-7.95 (m, 4H, Ar-H) ppm. Anal. Calcd. for C14H11ClN4OS2 (350.84): C, 47.93; H, 3.16; N, 15.97. Found: C, 47.89; H, 3.21; N, 16.01.5-amino-4-(4-chlorophenyl)-2-ethoxythieno[2,3-d]pyrimidine-6-carboxamide (19). A mixture of 18 (0.01 mol) and 50 ml ethoxide solution (prepared by dissolving 0.01 mol of Na in 50 ml absolute ethanol) was refluxed for 1h, cooled and poured onto cold water. The solid formed was filtered off, washed with water then dried and recrystallized from EtOH to give 19 as yellow crystals. m.p. 210-212 oC, Yield: 48%. IR (KBr) υmax: 3313, 3275, 3219, 3178 (NH2, CONH2), 2972 (CH), 1656 (C=O), 1593 (C=N) cm-1. 1H NMR (DMSO-d6) δ: 1.35 (t, 3H, CH3), 4.44 (q, 2H, CH2), 6.16 (s, br., 2H, NH2), 7.20 (s, br., 2H, NH2), 7.60-7.68 (m, 4H, Ar-H) ppm. Anal. Calcd. for C15H13ClN4O2S (348.81): C, 51.65; H, 3.76; N, 16.06. Found: C, 51.69; H, 3.71; N, 16.02.9-(4-chlorophenyl)-7-ethoxypyrimido[4',5':4,5]thieno[2,3-d]pyrimidin-4(3H)-one (20). A mixture of 19 (0.01 mol) and triethyl orthoformate (10 ml) in acetic anhydride (20 ml) was refluxed for 4h then cooled and poured into cold water. The precipitate formed was filtered off, dried and recrystallized from n-butanol to give 20 as yellow crystals. m.p. > 300 oC, Yield: 54%. IR (KBr) υmax: 3440 (NH), 3061, 2981 (CH), 1682 (C=O), 1583(C=N) cm-1. 1H NMR (DMSO-d6) δ: 1.39 (t, 3H, CH3), 4.50 (q, 2H, CH2), 7.53-7.88 (m, 4H, Ar-H), 8.16 (s, 1H, CH-pyrimidine), 12.90 (s, br., 1H, NH) ppm. Anal. Calcd. for C16H11ClN4O2S (358.81): C, 53.56; H, 3.09; N, 15.61. Found: C, 53.61; H, 3.05; N, 15.64.
4. Conclusions
- Using 6-aryl-5-cyano-2-thiouracil derivative as a precursor, several novel pyrimidines were synthesized. The antibacterial screening for some of the synthesized compounds indicated that all the tested compounds have antibacterial activities on the tested microorganisms.