-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2013; 3(1): 9-15
doi:10.5923/j.ajoc.20130301.02
Novel Synthesis and Antimicrobial Evaluation of Some New Cyclic Ketones
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAisha Hossan
Faculty of Education of Girls, King Khaled University, Abha, Saudi Arabia
Correspondence to: Aisha Hossan , Faculty of Education of Girls, King Khaled University, Abha, Saudi Arabia.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
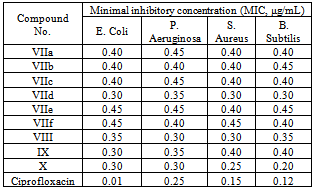
A series of 5-oxohexannitrile derivatives IIa-d was prepared by reaction of acrylonitrile with ketones Ia-d. On the other hand, semicarbazone derivatives IVa-d were obtained upon reaction of IIa-d with semicarbazide. Hydrolysis and esterification in one step reaction of δ-ketonitrile IIa-d resulted in the formation of the corresponding δ-ketoesters Va-d. The δ-ketoesters Va-d is readily cyclized to the corresponding cyclohexan-1,3-diones VIa-d when heated with alcoholic sodium methoxide. Moreover, coupling of VIa, VIb and VId with aryl diazonium chloride afforded the corresponding 2-(aryl diazenyl) derivatives VIIa-f, respectively. Furthermore, bis-(2,6-diketo-3-phenylcyclohexyl)methane VIII was synthesized by condensing VIa with benzaldehyde. Bromination of VId in dilute acetic acid afforded the corresponding bromo derivative IX. Heating of cyclohexanone-2-methyl propionate in alcoholic sodium methoxide afforded bicycle[1,3,3]nonan-2,9-dione X. These compounds were characterized by analytical and spectral analyses and screened for their antibacterial activity against Gram-possitive bacteria and Gram-negative bacteria. The synthesized compounds (VIIa-f)-X showed significant antibacterial activity against P. Aeruginosa (MIC 0.30-0.45 μg/mL), S. Aureus (MIC 0.25-0.45 μg/mL) and B. Subtilis (MIC 0.20-0.45 μg/mL) and exhibited moderate antibacterial activity against E. Coli (MIC 0.30-0.45 μg/mL) compared with the standard drug Ciprofloxacin.
Keywords: Cyclohexan-1,3-Dione, Semicarbazone, Cyclic Ketones, Antimicrobial Activity, Minimum Inhibitory Concentration (MIC)
Cite this paper: Aisha Hossan , Novel Synthesis and Antimicrobial Evaluation of Some New Cyclic Ketones, American Journal of Organic Chemistry, Vol. 3 No. 1, 2013, pp. 9-15. doi: 10.5923/j.ajoc.20130301.02.
Article Outline
1. Introduction
- The use of cyano compounds in organic synthesis is now receiving considerable interest[1, 2]. Our group has been involved in the last years in a program aiming to develop efficient procedures for the synthesis of polyfunctionally substituted azoles[3, 4], azines[5, 6] and their condensed derivatives[7, 8] utilizing simple and readily obtainable polyfunctionally substituted nitriles.
2. Results and Discussion
2.1. Chemistry
- The synthesis of cyclohexan-1,3-dione derivatives using acrylonitrile as starting material has now been achieved and reported herein. Thus, it has been found that when acrylonitrile was treated with ketones Ia-d in the presence of alkaline medium afforded the corresponding 5-oxohexannitrile derivatives IIa-d in good yield. The IR spectra of IIa-d, in general, showed absorption bands at 1730-1710 cm-1 corresponding to CO function and 2220 cm-1 due to C≡N group. The 1H-NMR spectrum of IIa revealed a singlet signal at δ 2.11 ppm due to CH3 protons, a triplet signal at δ 2.41 ppm attributable to COCH2 protons, two multiplet signals at δ 1.94 and 1.87 ppm due to CH2CH2CN protons. The 1H-NMR spectra of IIb-d showed characteristic signals, so compound IIb showed two singlet signals at δ 1.11 and 1.99 ppm corresponding to 2CH3 groups, while IIc showed signals due to the n-propyl group (n-C3H7) at δ 0.9 (s, CH3), 1.33 (m, CH2) and 1.53 ppm (q, CH2), finally, the 1H-NMR spectrum of IId showed the aromatic protons at δ 7.29-7.45 ppm as multiplet signals. Structure II was further confirmed by its conversion to the corresponding semicarbazone derivative IV. The IR spectra of the semicarbazone derivatives IVa-d, in general, showed absorption bands at 3420-3350 cm-1 due to NH2 and NH functions, while, their 1H-NMR spectra showed two singlet exchangeable signals at δ 6.06 and 7.09 ppm corresponding to NH2 and NH protons. Hydrolysis and esterification in one step reaction of δ-ketonitriles IIa-d resulted in the formation of the corresponding δ-ketoesters Va-d. Structure V was established based on spectral analysis. So, the IR spectra of Va-d showed in general, well defined bands due to carbonyl functions (ester at 1720 cm-1 and ketonic at 1695 cm-1). The 1H-NMR spectrum of Va revealed two singlet signals at δ 2.11 and 3.66 ppm due to protons of two methyl groups (COCH3 and COOCH3, respectively), two triplet signals at δ 2.14, 2.31 and multiplet at δ 2.4 ppm attributable to three CH2 groups, its mass spectrum showed the molecular ion peak at m/z = 144 (M+, 27%).The δ-ketoesters Va-d is readily cyclized to the corresponding cyclohexan-1,3-dione derivatives VIa-d when heated with alcoholic sodium methoxide (Scheme 1). Structures VIa-d were elucidated by correct analytical and spectral data. The IR spectra showed in general well defined absorption bands at 1735 cm-1 due to cyclic C=O group. The 1H-NMR spectrum of VIb showed two signals, one of them is doublet at δ 1.11 ppm and the other is multiplet at δ 2.52 ppm attributable to CH3 and CH protons at C4, respectively. The COCH2CO protons appeared as doublet of doublet at δ 3.56: 3.66 ppm and two multiplet signals at δ 1.94: 1.64 and 2.4: 2.5 ppm for other two CH2 groups, while its mass spectrum showed the molecular ion peak at m/z = 126 (M+). The 1H-NMR spectra of VIc, d showed characteristic signals corresponding to the alkyl and aryl groups at position-4. So, the n-propyl group at position-4 in VIc appeared as triplet signal for CH3 protons at δ 0.9 ppm, quartet signal at δ 1.53 ppm for CH2 protons and at δ 1.33 ppm as multiplet for the other CH2 protons, while the benzene ring protons in VId appeared at δ 7.29-7.51 ppm as multiplet signals.< Scheme 1 >
 | Figure 1. Structures of 2-arylazo of cyclohexane-1,3-dione derivatives VIIa-f |
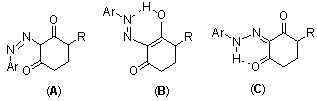 | Figure 2. The tautomeric structures of compounds VIIa-f |
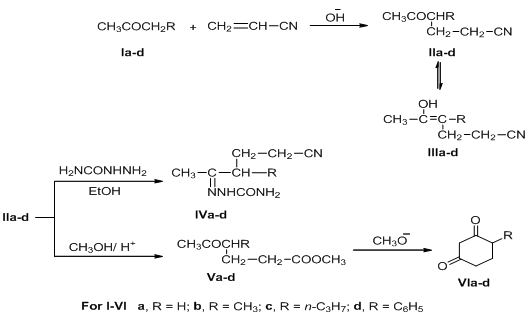 | Scheme 1. Synthetic routes for 4-substituted-cyclohexane-1,3-dione VIa-d |
|
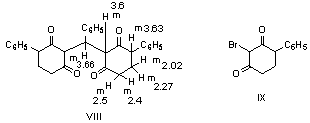 | Figure 3. Structures of phenylbis-(2,6-diketo-3-phenyl cycloh yl)methane (VIII) and 2-bromo-4-phenyl cyclohexan-1,3-dione (IX) |
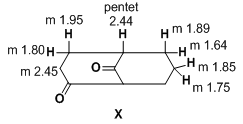 | Figure 4. Structure of (5R)bicyclo-[1.3.3]nonan-2,9-dione (X) |
2.2. Pharmacology
2.2.1. Antimicrobial Evaluation
- The reference standard Ciprofloxacin inhibited Gram negative bacteria E. Coli and P. Aeruginosa at a MIC of 0.01 μg/mL and 0.25 μg/mL, respectively, whereas, against Gram positive bacteria S. Aureus and Bacillus Subtilis MIC was found to be 0.15 μg/mL and 0.12 μg/mL, respectively.
|
3. Experimental
3.1. Instruments
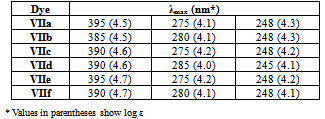
- All melting points are recorded on Gallenkamp electrothermal melting point apparatus. The IR spectra were recorded for KBr disc on a Mattson 5000 FTIR spectrophotometer. The 1H-NMR spectra were measured on a Bruker AC 300 (300 MHz) in DMSO-d6 as solvent, using TMS as an internal standard, and chemical shifts are expressed as δppm. The mass spectra were determined on Finnigan Incos 500 (70 ev). Elemental analyses were carried out at the Microanalytical Unit of the Faculty of Science, Cairo University, Giza, Egypt.Synthesis of nitrile derivatives IIa-dGeneral procedure: Sodium metal (0.46 g, 0.02 mol) is dissolved in the appropriate ketone Ia-d (0.7 mol) at 90℃. Then the solution was cooled to 0℃ and acrylonitrile (28 g, 0.52 mol) is added dropwise within 20 min at 50-60℃. The reaction mixture was stirred for 15 min, then cooled and neutralized with glacial acetic acid (3 mL), and extracted with ether; the ethereal extract was dried and the solvent evaporated under vacuo to give compounds IIa-d in good yields.5-Oxohexanenitrile (IIa):[19, 20]Yield (60%); b.p. 110-115 ℃ / 16 torr (Lit.[20], b.p. 112 ℃ / 14 torr); IR (Nojel): ν/cm-1= 2220 (C≡N), 1725 (CO); 1H-NMR(DMSO-d6) δ (ppm): 2.11 (s, 3H, CH3), 2.41 (t, 2H, CH2), 1.94 (m, 2H, CH2CN), 1.87 (m, 2H, CH2).4-Methyl-5-oxohexanenitrile (IIb):[21]Yield (53%); b.p. 110 ℃ / 12 torr (Lit.[21], b.p. 115 ℃ / 15 torr); IR (Nojel): ν/cm-1= 2220 (C≡N), 1730 (CO); 1H-NMR(DMSO-d6) δ (ppm): 1.11 (s, 3H, CH3), 1.99 (t, 2H, CH3CO), 2.0 (q, 2H, CH2), 2.41 (t, 2H, CH2), 2.52 (m, 2H, CH2).4-Acetylheptanenitrile (IIc):[22]5-Oxo-4-phenylhexanenitrile (IId): B.p. 160℃/ 10 torr (Lit.[23] b.p. 170-172℃/ 12 torr).Semicarbazone (IVa):[24] Yield (52%); m.p. 128℃; IR (KBr): ν/cm-1= 3420-3315 (NH2), 3150 (NH), 1675 (CO); 1H-NMR(DMSO-d6) δ (ppm): 2.0 (m, 2H, CH2), 1.91 (m, 2H, CH2), 2.10 (s, 3H, CH3), 2.4 (t, 2H, CH2), 6.06 (s, 2H, NH2), 7.09 (s, 1H, NH).Semicarbazone (IVb): Yield (45%); m.p. 130℃ (Lit.[21] m.p. 132℃).Semicarbazone (IVc): Yield (50%); m.p. 141℃ (Lit.[23] m.p. 163-165℃); IR (KBr): ν/cm-1= 3400-3310 (NH2), 3300 (NH), 1680 (CO); 1H-NMR(DMSO-d6) δ (ppm): 0.9 (t, 3H, CH3), 1.30 (m, 2H, CH2), 1.31 (m, 2H, CH2), 1.4 (m, 1H, CH), 1.41 (q, 2H, CH2), 1.71 (q, 2H, CH2), 1.95 (s, 3H, CH3), 6.1 (s, 2H, NH2), 7.2 (s, 1H, NH).Semicarbazone (IVd): m.p. 160℃ (Lit.[21] m.p. 163-165℃).Methyl-4-phenyl-4-acetylbutirate (Vd)A mixture of IId (0.4 mol) and 200 mL anhydrous methanol saturated with dry HCl gas was stirred, left to stand overnight, then cooled to 0℃, diluted with water and extracted with benzene. The organic layer washed with water and then with 5% sodium bicarbonate solution and finally with water. The solvent evaporated under vacuum to give Vd. Yield (60%); b.p. 142-144 / 4 mm (Lit.[23] b.p. 112-114 / 0.1 mm); IR (Nojel): ν/cm-1= 1720 (CO ester), 1695 (CO); 1H-NMR(DMSO-d6) δ (ppm): 2.13 (s, 3H, CH3), 2.25 (m, 2H, CH2), 2.35 (m, 2H, CH2COO), 3.65 (t, H, COCH), 3.70 (s, 3H, COOCH3), 7.29-7.40 (m, 5H, Ar-H). ηD20 1.5078; d429 1.0814.Methyl 5-oxohexanoate (Va)It was prepared by the above method. b.p. 85-89 / 7 mm (Lit.[24] b.p. 85-89 / 7 mm); 1H-NMR(DMSO-d6) δ (ppm): 2.11 (s, 3H, CH3), 2.14 (t, 2H, CH2), 2.31 (t, 2H, CH2COO), 2.40 (m, 2H, COCH2), 3.66 (s, 3H, COOCH3). ηD20 1.4269; d429 1.0267. MS (m/z, %) = 144 (M+, 10).Methyl 4-methyl-5-oxo-hexanoate (Vb)It was prepared by the above method. b.p. 90-91 / 6 mm (Lit.[25] b.p. 90-91 / 6 mm); 1H-NMR(DMSO-d6) δ (ppm): 1.11 (s, 3H, CH3), 1.90 (s, 3H, CH3CO), 1.98 (m, 2H, CH2), 2.35 (t, 2H, CH2COO), 2.52 (t, 1H, COCH), 3.65 (s, 3H, COOCH3). ηD20 1.4325; d429 1.0096. MS (m/z, %) = 158 (M+, 15).Methyl 4-acetylheptanoate (Vc)It was prepared by the above method. b.p. 95-100 / 6 mm (Lit.[26] b.p. 95-100 /6 mm); 1H-NMR(DMSO-d6) δ (ppm): 0.9 (t, 3H, CH3), 1.33 (m, 2H, CH2), 1.53 (m, 2H, CH2), 1.91 (s, 3H, CH3CO), 1.97 (m, 2H, CH2), 2.31 (m, 1H, COCH), 2.40 (m, 2H, CH2COO), 3.66 (s, 3H, COOCH3). ηD20 1.4380; d429 0.9773. MS (m/z, %) = 186 (M+, 28).4-Phenylcyclohexan-1,3-dione (VId)To a solution of sodium methoxide (0.05 mol, 20 mL methanol) in methanol was added Vd (0.05 mol) and refluxed with continuous stirring. The reaction mixture was cooled, neutralized with H2SO4 (30 mL, 2N), excess methanol removed under vacuum. To the residue 10 mL water was added and extracted with ether (30 mL). The extract was worked up with 20% cold sodium hydroxide and then acidified with 20% H2SO4 to give the diketone as oily material. The oily product extracted with ether again, dried (sodium sulfate) and the solvent was removed under vacuum. The resulting oily residue was triturated with acetone to give VId as a solid. Yield (60%); m.p. 112℃; IR (KBr): ν/cm-1= 1735 (CO); 1H-NMR(DMSO-d6) δ (ppm): 1.69: 1.94 (m, 2H, CH2), 2.4: 2.5 (m, 2H, CH2), 3.52 (m, 1H, CH), 3.56:3.66 (d.d., COCH2CO), 7.29-7.51 (m, 5H, Ar-H). MS (m/z, %) = 188 (M+, 70).Cyclohexan-1,3-dione (VIa). It was prepared by the above method from Va as yellow oil which solidified on cooling for long time, m.p. 102oC; yield 75%.4-Methyl cyclohexan-1,3-dione (VIb)It was prepared by the above method from Va as yellow oil. b.p. 140-145 / 9 mm (Lit.[27] b.p. 111 /0.025 mm); 1H-NMR(DMSO-d6) δ (ppm): 1.11 (d, 3H, CH3), 1.69: 1.94 (m, 2H, CH2), 2.40: 2.51 (m, 2H, CH2), 2.53 (m, 1H, CH), 3.56: 3.66 (d.d., 2H, COCH2CO). MS (m/z, %) = 126 (M+, 35).4-Propyl cyclohexan-1,3-dione (VIc)It was prepared by the above method from Vc; m.p. 75-76℃ (from ether); yield (60%); 1H-NMR(DMSO-d6) δ (ppm): 0.9 (t, 3H, CH3), 1.33 (m, 2H, CH2), 1.53 (q, 2H, CH2), 1.69: 1.94 (m, 2H, CH2), 2.34 (m, 1H, CH), 2.41: 2.50 (m, 2H, CH2), 3.56: 3.66 (d.d., 2H, COCH2CO). MS (m/z, %) = 154 (M+, 63).2-Phenylazocyclohexan-1,3-dione (VIIa)To a solution of cyclohexan-1,3-dione (VIa) (0.05 mol) in 10% sodium hydroxide (10 mL) was added dropwise with continuous stirring a cold solution of benzene diazonium chloride (0.15 g, 7 mL HCl, 0.1 g sodium nitrite). After cooling for 2 h, the reaction mixture was left to stand at room temperature overnight, when a yellow solid was obtained, m.p. 130℃ (from ethanol); UV λmax (methanol) 395, 275, 248 nm; log ε 4.5, 4.1, 4.3, respectively (Lit.[27] m.p. 135℃).2-(p-Methyl phenyl)azocyclohexan-1,3-dione (VIIb)It was prepared by the above method from p-tolyl diazonium chloride and VIa; m.p. 172℃; yield (80%); IR (KBr): ν/cm-1= 3150 (NH), 1610-1680 (CO), 1500 (C=N); UV λmax: 385, 280, 248 nm; log ε 4.5, 4.1, 4.3, respectively. Analysis for C13H14N2O2: Calc. C, 67.81; H, 6.13; N, 12.17%. Found: C, 67.77; H, 6.10; N, 12.09%. MS (m/z, %) = 230 (M+, 100).4-Methyl-2-phenylazocyclohexan-1,3-dione (VIIc)It was prepared by the above method from benzene diazonium chloride and VIb; m.p. 116℃; yield (50%); IR (KBr): ν/cm-1= 3155 (NH), 1675 (CO), 1550 (C=N); UV λmax: 390, 275, 248 nm; log ε 4.6, 4.2, 4.2, respectively. Analysis for C13H14N2O2: Calc. C, 67.81; H, 6.13; N, 12.17%. Found: C, 67.79; H, 6.09; N, 12.10%. MS (m/z, %) = 230 (M+, 100).4-Methyl-2-(p-nitrophenylazo)cyclohexan-1,3-dione derivatives (VIId)It was prepared by the above method from p-nitrophenyl diazonium chloride and VIb; m.p. 157℃; yield (62%); IR (KBr): ν/cm-1= 3250 (NH), 1680 (CO), 1530 (NO2), 1500 (C=N), 1350 (NO2); UV λmax: 390, 285, 245 nm; log ε 4.6, 4.0, 4.1, respectively. Analysis for C13H13N3O4: Calc. C, 56.72; H, 4.76; N, 15.27%. Found: C, 56.66; H, 4.71; N, 15.19%. MS (m/z, %) = 275 (M+, 100).4-Phenyl-2-phenylazocyclohexan-1,3-dione (VIIe)It was prepared by the above method from benzene diazonium chloride and VId; m.p. 130℃; yield (80%); IR (KBr): ν/cm-1= 3150 (NH), 1675 (CO), 1510 (C=N); UV λmax: 395, 275, 248 nm; log ε 4.7, 4.2, 4.2, respectively. Analysis for C18H16N2O2: Calc. C, 73.95; H, 5.52; N, 9.58%. Found: C, 73.89; H, 5.48; N, 9.49%. MS (m/z, %) = 292 (M+, 100).4-Phenyl-2-(p-methoxyphenylazo)cyclohexan-1,3-dione (VIIf)It was prepared by the above method from p-methoxyphenyl diazonium chloride and VId; m.p. 177℃; yield (65%); IR (KBr): ν/cm-1= 3150 (NH), 1680 (CO), 1500 (C=N); UV λmax: 390, 280, 248 nm; log ε 4.7, 4.1, 4.1, respectively. Analysis for C19H18N2O3: Calc. C, 70.79; H, 5.63; N, 8.69%. Found: C, 70.71; H, 5.59; N, 8.78%. MS (m/z, %) = 322 (M+, 100).Phenylbis-(2,6-diketo-3-phenylcyclohexyl)methane (VIII). To a solution 25 mL, 4% of VId in ethanol (50 mL, 50%) was added 5 drops benzaldehyde. The reaction mixture was refluxed for 0.5 h then left to stand at room temperature for three days to give a solid product VIII; m.p. 160℃; yield (55%); IR (KBr): ν/cm-1= 1730 (CO); 1H-NMR(DMSO-d6) δ (ppm): 2.02: 2.27 (m, 4H, 2CH2), 2.40: 2.5 (m, 4H, 2CH2), 3.6 (m, 2H, 2CH), 3.63 (m, 2H, 2CH-Ph), 7.2-7.45 (m, 15H, Ar-H). MS (m/z, %) = 464 (M+, 100). Analysis for C31H28O4: Calc. C, 80.15; H, 6.08%. Found: C, 80.0; H, 5.95%. 2-Bromo-4-phenylcyclohexan-1,3-dione (IX)To VId (0.05 mol) in 200 mL cold water was added dropwise bromine (0.06 mol) and heated to obtain homogenous solution. The reaction mixture was cooled and the solid product was filtered and recrystallized from ethanol to give the bromo derivative IX; m.p. 151℃; 1H-NMR(DMSO-d6) δ (ppm): 2.01-2.50 (m, 4H, 2CH2), 3.63 (t, 1H, C4-H), 5.15 (s, 1H, C2-H), 7.2-7.4 (m, 5H, Ar-H). MS (m/z, %) = 265 (M+, 100). Analysis for C12H11BrO2: Calc. C, 53.96; H, 4.15%. Found: C, 53.81; H, 3.95%.(5R)Bicyclo-[1.3.3]nonan-2,9-dione (X)It was obtained by the general previous method for preparing compound V from 2-(2’-methoxycarbonyl ethyl) cyclohexanone (V); m.p. 141oC; IR (KBr): ν/cm-1= 1725 (CO); 1H-NMR(DMSO-d6) δ (ppm): 1.76-1.81 (m, 5H, CH + 2CH2), 2.04 (m, 2H, CH2), 2.21 (m, 2H, CH2), 2.45 (t, 2H, CH2), 3.08 (t, 2H, CH2). MS (m/z, %) = 152 (M+, 81). Analysis for C9H12O2: Calc. C, 71.03; H, 7.95%. Found: C, 71.0; H, 7.90%.
3.2. Antibacterial activity
- All the compounds were screened for their in vitro antibacterial activity against two Gram negative strains, i.e. Escherichia Coli (MTCC 40) and Pseeudomonas Aeruginosa (MTCC 2453) and two Gram positive strains, i.e. Bacillus Subtilis (MTCC 121) and Staphylococcus Aureus(MTCC 96).Antibacterial activity was assessed by serial two fold dilution technique[28]. Ciprofloxacin was used as a standard drug. All the compounds were dissolved in dimethyl sulfoxide to give a concentration of 10 μg mL-1. Double strength nutrient broth was used as a growth media. The stock solution was serially diluted to give concentration of 5.0-0.01 μg/mL in nutrient broth. The inoculums size was approximately 106 colony forming units (CFU/ mL). The inoculated tubes were incubated for 24 h, the inoculated culture tubes were macroscopically examined for turbidity. The culture tube showing turbidity (lower concentration) and the culture tube showing no turbidity (higher concentration) gave the minimum inhibitory concentration (MIC) for the compound. The MIC for the (VIIa-f)-X and the standard drug, i.e. Ciprofloxacin are given in Table 2.