-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2012; 2(6): 132-135
doi:10.5923/j.ajoc.20120206.02
Synthesis of Monoaryl Hydrazides via the BF3 Catalyzed Reaction of Diethyl Azodicarboxylate with Substituted Benzenes
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLGary W. Breton, Michael J. Grigalunas, Joshua S. Hughes
Department of Chemistry, Berry College, Mount Berry GA, 30149, USA
Correspondence to: Gary W. Breton, Department of Chemistry, Berry College, Mount Berry GA, 30149, USA.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
DiethylAzodicarboxylate (DEAD) reacted readily with sufficiently activated aromatic compounds in the presence of 25 mol% BF3•O(Et)2 as Lewis acid catalyst to afford monoarylhydrazides in reasonable-to-high yields (41-94%).
Keywords: Arenes, Azodicarboxylates, Electrophilic Aromatic Substitution
Cite this paper: Gary W. Breton, Michael J. Grigalunas, Joshua S. Hughes, Synthesis of Monoaryl Hydrazides via the BF3 Catalyzed Reaction of Diethyl Azodicarboxylate with Substituted Benzenes, American Journal of Organic Chemistry, Vol. 2 No. 6, 2012, pp. 132-135. doi: 10.5923/j.ajoc.20120206.02.
Article Outline
1. Introduction
- Monoarylhydrazides (1, Scheme 1) are important intermediates for the synthesis of a variety of biologically active heterocycles including indoles[1], oxadiazolones[2], and pyrazolones[3]. Thus, it is not surprising that the pursuit of new and improved methods for the synthesis of aryl hydrazides remains an active area of research[4].
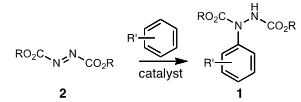 | Scheme 1. Synthesis of MonoarylHydrazides via Electrophilic Aromatic Substitution of Azodicarboxylates |
2. Results and Discussion
- In the few studies that reported the use of the Lewis acid BF3 to catalyze the EAS reactions of DEAD with aromatic compounds, one or more equivalents of the acid catalyst were utilized[12]. Indeed, the addition of one equivalent of BF3•O(Et)2 to an equimolar mixture of DEAD (2.5 mmol) and anisole in CH2Cl2 as solvent resulted in formation of the desired adduct 3a (Figure 1) in reasonable yield (64%). We were able to decrease the catalyst load to 25 mol% without affecting the product yield (64%) although the reaction required somewhat longer reaction time (1 h vs 0.5 h). In both cases, a disubstituted product was formed in addition to the desired monoadduct. This is not surprising given that the monoadductis expected to be electronically activated relative to starting anisole. Using 2 equivalents of anisole was enough to virtually eliminate the formation of the disubstituted product and the yield of 3a increased to 90%. We subsequently found that with other substrates, such as 1,3-dimethoxybenzene to form 3c (vide infra), upwards of 10 equivalents of the aromatic substrate were required to squelch formation of di- and polysubstituted products. Fortunately, however, in these cases unreacted aromatic starting material could be readily recovered (after quenching of the BF3 catalyst) via either direct distillation from the crude reaction mixture or column chromatography.
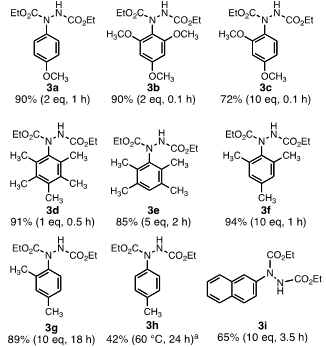 | Figure 1. Products of the EAS reactions of DEAD with various aromatic substrates. All reactions were conducted according to the General Procedure described in the experimental unless otherwise noted. Percent yields are for purified products. In parentheses are indicated equivalents of substrate, reaction time, and temperature if relevant. aToluene was used as the solvent in the reaction. The ratio of para:ortho adduct was ~ 70:30 as determined by 1H NMR integration |
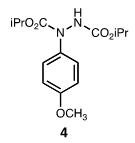 | Figure 2. Product of Reaction of Diisopropylazodicarboxylate with Anisole |
3.Experimental
3.1. General Procedure for the Reaction of Diethyl Azodicarboxylate (DEAD) with Substituted Benzenes
- To a stirring solution of aromatic substrate (1-10 equivalents as per Figure 1) in 8 mL CH2Cl2 was added 0.44 g (2.5 mmol) of DEAD as a solution in 2 mL CH2Cl2 followed by 80 μL (0.63 mmol, 25 mol%) of BF3•(Et)2O via syringe. The resulting solution stirred until the DEAD was completely consumed (as determined by TLC). The reaction mixture was washed with H2O (10 mL), and the resulting organic layer dried over Na2SO4, filtered, and concentrated.The product was purified via column chromatography (SiO2, using mixtures of EtOAc and hexanes as eluent).
3.1.1. Diethyl 1-(4-methoxyphenyl)hydrazine-1, 2-dicarboxylate (3a)
- According to the general procedure using 0.54 g (5 mmol, 2 eq) of anisole, 0.70 g (2.26 mmol, 90% yield) of 3a was isolated as a colorless viscous oil.1H NMR (60 MHz, CDCl3): 7.92 (br s, 1H, NH), 7.35 (d, 2H, J = 9.2 Hz), 6.81 (d, 2H, J = 9.2 Hz), 4.19 (q, 2H, J = 7.1 Hz), 4.18 (q, 2H, J = 7.1 Hz), 3.74 (s, 3H), 1.22 (br t, 6H, J = 7.1 Hz).13C NMR (14 MHz, CDCl3): 158.2, 156.6, 155.5, 135.0, 126.6, 113.9, 62.8, 62.0, 55.4, 14.4(2C).HRMS (EI) m/z[M+Na]+ calculated for C13H18N2NaO5: 305.1108. Found 305.1110.
3.1.2. Diethyl 1-(2,4,6-trimethoxyphenyl)hydrazine-1, 2-dicarboxylate (3b)
- According to the general procedure using 0.84 g (5 mmol, 2 eq) of 1,3,5-trimethoxybenzene, 0.77 g (2.25 mmol, 90% yield) of 3b was isolated as a white solid; mp 145-146℃.1H NMR (60 MHz, CDCl3): 7.09 (br s, 1H NH), 6.13 (s, 2H), 4.16 (q, 4H, J = 7.1 Hz), 3.83 (s, 6H), 3.80 (s, 3H), 1.25 (t, 3H, J = 7.1 Hz), 1.17 (br t, 3H, J = 7.1 Hz).13C NMR (14 MHz, CDCl3): 160.6(2C), 156.5, 155.4, 112.5, 90.7, 62.1, 61.2, 55.7(2C), 55.1, 14.1(2C).Anal. Calcd. For C15H22N2O7: C, 52.61; H, 6.48; N 8.19. Found: C, 52.85; H; 6.73, N, 8.24.
3.1.3. Diethyl 1-(2,4-dimethoxyphenyl)hydrazine-1, 2-dicarboxylate (3c)
- According to the general procedure using 3.45 g (25 mmol, 10 eq) of 1,3-dimethoxybenzene, 0.57 g (1.83 mmol, 73% yield) of 3c was isolated as a colorless viscous oil.1H NMR (60 MHz, CDCl3): 7.49 with 7.33 (br s, 1H, NH), 7.20 (br s, 1H), 6.55-6.35(m, 2H), 4.18 (br q, 4H, J = 7.1 Hz), 3.80 (s, 6H), 1.25 (t, 3H, J = 7.1 Hz), 1.21 (br t, 3H, J = 7.1 Hz).13C NMR (14 MHz, CDCl3): 160.6(2C), 156.2, 155.8, 130.2, 123.6, 104.1, 99.2, 62.6, 61.7, 55.5(2C), 14.4(2C).HRMS (EI) m/z[M+Na]+ calculated for C14H20N2NaO6: 335.1214. Found 335.1214.
3.1.4. Diethyl 1-(2,3,4,5,6-pentamethylphenyl)hydrazine-1, 2-dicarboxylate (3d)
- According to the general procedure using 0.37 g (2.5 mmol, 1 eq) of 1,2,3,4,5-pentamethylbenzene, 0.73 g (2.27 mmol, 91% yield) of 3d was isolated as a white solid; mp 179-181 ℃. 1H NMR (60 MHz, CDCl3):6.96 and 6.81 (br s, 1H, NH), 4.21 (q, 2H, J = 7.1 Hz), 4.17 (q, 2H, J = 7.1 Hz), 2.26 (s, 6H), 2.20 (s, 9H), 1.25 (t, 3H, J = 7.1 Hz), 1.20 (t, 3H, J = 7.1 Hz). 13C NMR (14 MHz, CDCl3): 156.5, 156.2, 136.9, 135.4, 133.5, 131.7, 62.8, 61.9, 16.9, 16.7, 15.7, 17.7, 14.4.Anal. Calcd. For C17H26N2O4: C, 63.32; H, 8.13; N 8.69. Found: C, 63.24; H, 8.36; N, 8.62.
3.1.5. Diethyl 1-(2,3,5,6-tetramethylphenyl)hydrazine-1, 2-dicarboxylate (3e)
- According to the general procedure using 1.68 g (12.5 mmol, 5 eq) of durene, 0.65 g (2.11 mmol, 85% yield) of 3e was isolated as a white solid; mp 185-186 ℃.1H NMR (400 MHz, CDCl3): 7.32 (br s, 1H, NH), 6.96 (br s, 1H) 4.20 (q, 2H, J = 7.1 Hz), 4.16 (q, 2H, J = 7.1 Hz), 2.22 (s, 12H), 1.23 (t, 3H, J = 7.1 Hz), 1.19 (t, 3H, J = 7.1 Hz). 13C NMR (14 MHz, CDCl3): 156.3(2C), 138.9, 134.6, 132.1, 131.5, 62.8, 61.9, 20.0, 14.7, 14.6, 14.4.Anal. Calcd. For C16H24N2O4: C, 62.30; H, 7.85; N 9.09. Found: C, 62.70; H, 8.02; N, 9.05
3.1.6. Diethyl 1-(2,4,6-trimethylphenyl)hydrazine-1, 2-dicarboxylate (3f)
- According to the general procedure using 1.50 g (12.5 mmol, 5 eq) of mesitylene, 0.69 g (2.35 mmol, 94% yield) of 3f was isolated as a white solid; mp 159-160 ℃ (lit.[12c] 159-161 ℃).1H NMR (60 MHz, CDCl3): 7.52 and 7.23 (br s, 1H, NH) 6.87 (br s, 2H), 4.14 (q, 2H, J = 7.1 Hz), 4.10 (q, 2H, J = 7.1 Hz), 2.34 (s, 6H), 2.26, s (3H), 1.22 (t, 3H, J = 7.1 Hz), 1.18 (t, 3H, J = 7.1 Hz).13C NMR (14 MHz, CDCl3): 156.47 (2C), 137.9, 136.1, 136.0, 129.3, 62.9, 61.9, 20.9, 18.2, 14.5, 14.4.
3.1.7. Diethyl 1-(2,4-dimethylphenyl)hydrazine-1, 2-dicarboxylate (3g)
- According to the general procedure using 0.43 g (25 mmol, 10 eq) of m-xylene, 0.62 g (2.21 mmol, 89% yield) of 3g was isolated as a white solid.;mp 83-84 ℃ (lit.[12d]81-82 ℃).1H NMR (60 MHz, CDCl3): 7.4-6.9 (m, 4H), 4.15 (q, 2H, J = 7.1 Hz), 3.13 (q, 2H, J = 7.1 Hz), 2.31 (s, 3H), 2.26 (s, 3H), 1.25 (t, 3H, J = 7.1 Hz), 1.22 (t, 3H, J = 7.1 Hz).13C NMR (14 MHz, CDCl3): 156.5, 155.7, 138.0, 137.6, 135.2, 131.1, 127.4, 127.0, 62.5, 61.6, 20.7, 17.3, 14.2(2C).Anal. Calcd. For C15H22N2O7: C, 59.97; H, 7.19; N 10.00. Found: C, 60.27; H, 7.49; N, 9.97.
3.1.8. Diethyl 1-(4-methylphenyl)hydrazine-1, 2-dicarboxylate (3h)
- According to the general procedure using 10 mL of toluene as solvent and heating the reaction mixture to 60 ℃ for 24 h, 0.28 g (1.05 mmol, 42% yield) of 3h was isolated as a colorless viscous oil. 1H NMR (60 MHz, CDCl3): δ(mixture of isomers) 7.8 and 7.66 (br s, 1H, NH), 7.39-7.00 (m, 4H), 4.15 (q, 2H, J = 7.1 Hz), 4.12 (q, 2H, J = 7.1 Hz), 2.30 (s, 3H), 1.23 (t, 6H, J = 7.1 Hz).13C NMR of para isomer (14 MHz, CDCl3): δ 156.6 (2C), 139.3, 136.2, 129.2, 124.4, 62.8, 62.1, 20.9, 14.4(2C). 13C NMR of ortho isomer (14 MHz, CDCl3): δ155.2(2C), 140.4, 135.6, 130.7, 128.2, 127.7, 126.6, 62.8, 62.1, 17.7, 14.4 (2C). HRMS (EI) m/z[M+Na]+ calculated for C13H18N2NaO4: 289.1159. Found 289.1160.
3.1.9. Diethyl 1-(naphthalen-2-yl)hydrazine-1, 2-dicarboxylate (3i)
- According to the general procedure using 1.60 g (12.5 mmol, 5 eq) of naphthalene, 0.49 g (1.63 mmol, 65% yield) of 3i was isolated as a white solid; 137 -138 ℃[10].1H NMR (60 MHz, CDCl3): δ 8.07-7.26 (m, 8H), 4.15 (q, 4H, J = 7.1 Hz), 1.25 (t, 3H, J = 7.1 Hz), 1.13 (br t, 3H, J = 7.1 Hz). 13C NMR (14 MHz, CDCl3): δ 156.7, 156.1, 138.0, 134.5, 130.2, 128.9, 128.4, 126.9, 126.3, 126.0, 125.7, 122.8, 63.1, 62.2, 14.5(2C). Anal. Calcd. For C16H18N2O4: C, 63.55; H, 6.00; N 9.27. Found: C, 63.71; H, 6.25; N, 9.19.
3.1.10. Diisopropyl 1-(4-methoxyphenyl)hydrazine-1, 2-dicarboxylate (4)
- According to the general procedure using 0.54 g (5 mmol, 2 eq) of anisole and 0.51 g (2.5 mmol) of diisopropylazodicarboxylate, 0.70 g (2.26 mmol, 90% yield) of 4 was isolated as a colorless viscous oil. Spectral data were in accord with literature values[4c,e].
4. Conclusions
- In summary, the reaction of DEAD with sufficiently activated aromatic compounds provides a general, highyield method for the synthesis of monoarylhydrazides. The optimized method requires only 25 mol% of the inexpensive and readily available Lewis acid catalyst BF3•O(Et)2. The greatest limitation is the requirement for aromatic rings at least as electronically activated as toluene.
ACKNOWLEDGEMENTS
- This material is based upon work supported by the National Science Foundation under CHE - 1125616. Generous financial support from the Berry College Faculty Development Grant Program is appreciated.