-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2012; 2(6): 127-131
doi:10.5923/j.ajoc.20120206.01
Stereoselective Synthesis of Bicyclic Lactones Via Annelation Protocol
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLBello Y. Makama
Department of Chemistry, Faculty of Science & Science Education, Kano University of Science & Technology. P.M.B. 3244 Wudil, Kano State, Nigeria
Correspondence to: Bello Y. Makama, Department of Chemistry, Faculty of Science & Science Education, Kano University of Science & Technology. P.M.B. 3244 Wudil, Kano State, Nigeria.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
A successful two-step annelation protocol of diesters and methyl bromoacetate with 2- chlorocyclopentanone derivatives was efficiently pursued, which gave suitably substituted bicyclic lactones in high overall yields and with complete streoselectivity mediated by K-Selectride and Wilkinsons catalyst, is reported. As part of a program aimed at rapid synthesis of bicyclic lactones which inherently occurs in many active natural products, this paper has shown a novel and rare methods for the synthesis of these important compounds based on alkylation of cyclopentanone derivatives and further demonstrate efficient reduction protocol of these compounds to the bicyclci lactones
Keywords: Annelation, Diesters, Methyl Bromo Acetate, Bicyclic Lactones
Cite this paper: Bello Y. Makama, Stereoselective Synthesis of Bicyclic Lactones Via Annelation Protocol, American Journal of Organic Chemistry, Vol. 2 No. 6, 2012, pp. 127-131. doi: 10.5923/j.ajoc.20120206.01.
Article Outline
1. Introduction
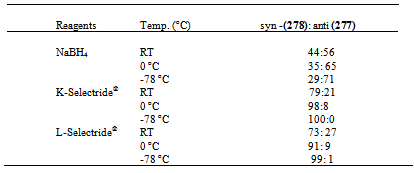
- Bicyclic lactones systems are among Nature’s preferred building blocks for the construction of tricyclic lactones of varied biological activities. In particular, the γ-butyrolactone moiety is a recurrent feature of many naturally occurring substances examples are brasoside and littoralissone.1,2 The synthetic approaches to this bicyclic lactones have been imaginative and numerous,3-8 there is a continued demand for efficient methods for the assembling of these key compounds which give especially high levels of stereoselectivity. In this report, it is reported some of our preliminary results in the establishment of an effective methodology for the construction of these important compounds. It was envisaged that reduction of substituted cyclopentanone derivatives using methods of alkylation described by Fumihinko9 would give the ester (276). Stereocontrolled reduction with Selectride® reagents and subsequent lactonization would furnish the cis-fused bicyclic ring system (278). It therefore remained to investigate the annelation of a diketone with methyl bromoacetate and later explore the cyclization protocol. The success of this synthetic methodology would require the viability of the condensation between the cyclopentanone derivatives with the diester and methyl bromoacetate. The most important step would now be the selective Selectride® mediated reduction of the ketone in compounds (276) and (283) to give the syn-alcohols which could readily ring closed into the desired bicyclic structures.
2. Results & Discussion
2.1. Exploration of Direct Formation of Dimethy 2-(2-Oxocyclopentyl) Malonate
- Following a report by Fumihinko9 that 2-chlorocyclopentanone (275) can be converted into dimethyl 2-(2-oxocyclopentyl) malonate (276) we obtained the corresponding diester (276) in 53% yield. The 1H NMR spectrum fully supported the proposed structure (276), displaying a doublet centred at δ 3.84 ppm (J 5.8 Hz) corresponding to a methine proton bonded to the diester, a pair of three proton singlets at δ 3.78 ppm and at δ 3.73 ppm for methyl esters and a one proton resonance at δ 2.97-2.88 ppm for the methine adjacent to the ketone. The presence of the ester groups and the ketone was manifested in the IR spectrum, with characteristic absorptions at, 1738 cm-1 and 1749 cm-1 respectively. The mass spectrum fully supported this with a molecular ion of m/z 214 Scheme 1.1.
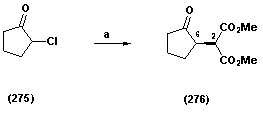 | Scheme 1.1. (a) dimethyl malonate, DMF/THF (1:1), NaH, 53% |
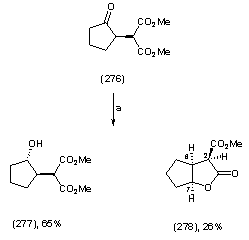 | Scheme 1.2. (a) ethanol, NaBH4, 0℃, 30 min |
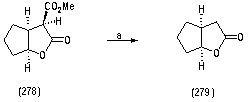 | Scheme 1.3. (a) silica gel, hex : ethyl acetate (1:3), 32% |
|
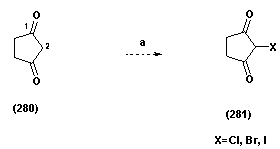 | Scheme 1.4. (a) NBS, NCS, KIO3, THF |
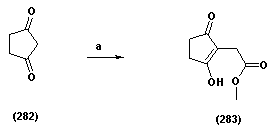 | Scheme 1.5. (a) MBA, Et3N, CH3CN, pH 2, 78% |
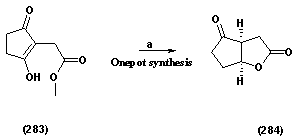 | Scheme 1.6. (a) Wilkinsons Catalyst, Catecholborane, aq. HCl, pH 2, 73% |
3. Experimental Techniques
- Commercial reagents were obtained from Aldrich and Lancaster chemical suppliers and were used directly as supplied or purified prior to use following the guidelines of Perrin and Amarego.15 Dichloromethane and acetonitrile were refluxed over and distilled from CaH2 prior to use. Diethyl ether and ethanol were obtained dry from Aldrich. THF was dried by distillation from the sodium benzophenone ketyl radical under nitrogen. Light petroleum is the fraction of petroleum ether boiling in the range 30-40 oC, and it was fractionally distilled through a 36 cm Vigreux column before use. Non-aqueous reagents were transferred under argon via syringe. Organic solutions were concentrated under reduced pressure on a Büchi rotary evaporator using a water bath. Thin-layer chromatography (TLC) was performed on Merck aluminium-backed plates coated with 0.2 mm silica gel 60-F plates. Visualization of the developed chromatogram was performed by UV fluorescence quenching at 254 nm, or by staining with a KMnO4 solution. 1H and 13C NMR spectra were recorded on a Bruker DPX250 (250 MHz for protons) and a Brüker AMX400 (400 MHz for protons). Data for 1H NMR are reported as follows: chemical shift (δ-ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), integration, coupling constant in (Hz). Data for 13C NMR spectra are reported in terms of chemical shift (ppm) down field from TMS. IR spectra were recorded on a Perkin Elmer Paragon 1000 or a Perkin Elmer 881 spectrometer as a thin film between sodium chloride plates or as a KBr disk. All absorptions are reported in terms of frequency of absorption (cm-1). Mass spectrometric data were recorded on VG Autospec, under conditions of chemical ionisation (C.I) using ammonia as the ionising source. Peaks are quoted in the form (m/z) (relative intensity).
3.1. Experimental Procedures
- Dimethyl 2-(2-oxocyclopentyl)malonate (276) To a stirred solution of NaH (209 mg, 5.49 mmol, 1.30 equiv) in THF-DMF (10 mL, 1:1) at 0 oC was added dimethyl malonate (724 mg, 5.49 mmol, 1.3 equiv) and the solution was stirred for 30 min at room temperature. 2-chlorocyclopentanone (275) (500 mg, 4.22 mmol, 1.00 equiv) was added to the solution and then further stirred at room temperature for 8 h, by which time TLC analysis, revealed the formation of a new product. Saturated aqueous NH4Cl solution (10 mL) was added to the mixture and the organic layer was extracted with ether (4 x 15 mL). The combined organic layers were washed with sat. NaHCO3 solution (4 x10 mL), brine (4 x 10 mL), dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel using a gradient of solvents, hexane: ethyl acetate from (5:1-100% ethyl acetate) to afford the title compound as a colourless oil (192 mg, 53%); υmax (thin film/cm-1), 2961, 1749, 1738; δH (250 MHz, CDCl3) 3.84 (1H, d, J 5.8 Hz, CH(CO2CH3)2), 3.78 (3H, s, CH3), 3.73 (3H, s, CH3), 2.97-2.88 (1H, m, CHC=O), 2.56-1.99 (4H, m, CH2CH, CH2C=O), 2.00-1.81 (2H, m, CH2CH2C=O); δC (62.5 MHz, CDCl3), 216.2, 169.1, 168.6, 53.9, 51.6, 51.2, 41.2, 38.2, 23.8 20.8; m/z (C.I) 215 (MH+, 100%), 214 (4%), 183 (11%), 155 (7%), 123 (3%), C10H15O5, requires 215.092, found , 215.0913Dimethyl -2,2-hydroxycyclopentyl)malonate (277)Methyl-2-oxo-hexahydro-2H-cyclopenta[b]furan-3-carboxylate (278)To a stirred solution of dimethyl2-(2-oxocyclopentyl)malonate (276) (80 mg, 0.37 mmol, and 1.00 equiv) in ethanol (7 mL) in an Erlenmeyer flask was added NaBH4 at room temperature (16.8 mg, 0.44 mmol, 1.20 equiv) in small portions over a period of 15 min The reaction mixture was stirred for further 30 minutes and then poured into water (5 mL). (5%) dilute hydrochloric acid (4 drops) was added and the organic layer was extracted with ether (2 x 10 mL), dried over MgSO4 and concentrated in vacuo. Column chromatography on silica, eluting with hexane : ether (3:1) afforded (277) as a colourless oil (66 mg, 65%); υmax (thin film/cm-1), 3504, 2955, 2255, 1731; δH (250 MHz, CDCl3) 3.96-3.91 (1H, m, CHOH), 3.68 (3H, s, CH3), 3.67 (3H, s, CH3), 3.32 (1H, d, J 7.8 Hz, CH(CO2Me)2), 2.33-2.28 (1H, m, CH), 1.88-1.83 (2H, m, CH2), 1.63-1.53 (3H, m, CH2), 1.28-1.25 (1H, m, CH2); δC (62.5 MHz, CDCl3) 169.2, 168.6, 76.2, 55.9, 51.5, 51.0, 46.8, 33.4, 28.3, 21.3; m/z (C.I) 216 (MH+, 9%), 159 (10%), 150 (4%), C10H17O5, requires 216.0998, found, 216.0935; further elution yielded (278) (18 mg, 26%); υmax (thin film/cm-1), 2957, 1742, 1733, ; δH (250 MHz, CDCl3) 5.06 (1H t, J 5.3 Hz, CHO), 3.73 (3H, s, CO2CH3), 3.68 (1H, d, J 3.3, CHC=O), 3.14-3.10 (1H, m, CHCHC=O), 1.98-1.56 (6H, m, CH2); δC (62.5 MHz, CDCl3) 176.9, 168.5, 86.5, 54.8, 53.7, 43.5, 33.5, 32.9, 23.9; m/z (C.I) 185 (MH+, 45%), 141 (51%), 134 (43%), 124(67), 101 (77%) 84 (54%) C9H13O4, requires 185.0814, found, 185.0806Hexahydrocyclopenta[b]furan-2-one (279) Methyl-2-oxo-hexahydro-2H-cyclopenta[b]furan-3-carboxylate (278) (50 mg, 0.27 mmol, 1.00 equiv) was subjected to column chromatography on silica eluting Hex : ethyl acetate (1:3) to afford the title compound (279) as a colourless oil (11 mg, 32%); υmax (thin film/cm-1), 2962, 2879, 1769, 1205, 982; δH (250 MHz, CDCl3) 5.02 ( 1H t, J 5.0 Hz, CHO), 2.97-2.80 (1H, m, CH2COO), 2.79 (1H, d, J 10 Hz, CH2COO), 2.28-2.15 (1H, m, CHCHO), 2.09-2.02 (1H, m, CH2), 1.97-1.80 (5H, m, CH2); δC (62.5 MHz, CDCl3) 177.2, 86.6, 37.8, 36.0, 33.5, 29.9, 23.4; m/z (C.I) 125 (MH-, 56%), 124 (58%), 109 (8%), 124 (58), 109 (8%) 107 (9%) C7H11O2, requires 125.0603, found, 125.062Methyl 2-(2-hydroxy-5-oxocyclopent-1-enyl)acetate(283)To a stirred solution of 1,3-cyclopentanedione (279) (5 g, 0.05 mmol, 1.00 equiv) in acetonitrile (80 mL) at 0 oC was added triethylamine (7.6 g, 0.07 mmol, 1.30 equiv), and the solution was stirred for 10 min at room temperature. Methyl bromoacetate (9.9 g, 0.07 mmol, 1.30 equiv) was then added and stirring was continued over night by which time TLC revealed a new spot. Saturated aqueous NH4Cl solution (15 mL) was added to the mixture and the organic layer was extracted with ethyl acetate (3 x 20 mL). The combined organic layers were washed with sat. NaHCO3 solution (2 x15 mL), brine (2 x 15 mL), dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel using a solvent gradient hexane : ethyl acetate from (4:1) to afford the title compound as a white powder (6.6 g, 78%); m.p. (148-151 oC); υmax (KBR/cm-1), 3389, 2927, 1726, 1656, 1574; δH (250 MHz, CDCl3) 4.61 (2H, s, CH2CO2CH3), 3.38 (3H, s, COOCH3), 2.72 (2H, t, J 2.5 Hz, CH2C=O), 2.49 (2H, t, J 2.5 Hz, CH2COH) ; δC (62.5 MHz, CDCl3) 205.8, 189.4, 167.5, 106.7, 67.9, 53.1, 34.8, 28.6; m/z (C.I) 171 (MH+, 100%), 170 (10%), 142 (14%), 131 (5%), 81 (2%) C8H11O4, requires 171.0657, found, 171.0657Tetrahydro-cyclopenta[b]furan-2,4-dione (284)To a stirred solution of Wilkinsons catalyst (RhPPh3Cl) (236.00 mg, 0.589 mmol, 0.5 equiv) in anhydrous THF (7 mL) under N2 cooled to -20 oC was added to methyl 2-(2-hydroxy-5-oxocyclopent-1-enyl)acetate (283) (200.0 mg, 1.18 mmol, 1.00 equiv), followed by the addition of catecholborane (424.45 mg, 3.54 mmol, 3.0 equiv). The reaction was allowed to stir for 24 hr, by which time TLC analysis revealed a new product been formed. The reaction was quenched by the addition phosphate buffer pH 7.00, (4 mL). The product was extracted with ether (4 x 10 mL), washed with 10% aqueous sodium sulfite (2 x 5 mL) and saturated sodium carbonate (4 x 5mL). The ether layer was followed by the addition of dilute hydrochloric acid (5%) at 0 oC until the pH 2 was reached, by which time, TLC analysis showed a new product has been formed. The aqueous layer was extracted with ether (4 x 10 mL), the extract dried over MgSO4 and concentrated in vacuo. Column chromatography on silica, eluting with hexane : ethyl acetate gave the product as a colourless oil (122 mg, 73%); υmax (thin film/cm-1), 2929, 2851, 1751, 1737, 1644, 1088; δH (250 MHz, CDCl3) 5.24 (1H, t, J 5.0 Hz, CHO), 2.96 (1H, dddd, J 5.0 Hz, J 2.5 Hz, CH=CH2C=O), 2.85 (1H, d, J 10 Hz, J 2.5 Hz, CH2C=O), 2.78 (1H, d, J 15.0 Hz, CH2C=O), 2.57-2.43 (3H, m, CH2CHO, CH2C=O), 2.32-2.22 (1H, m, CH2CHO); δC (62.5 MHz, CDCl3) 209, 176.6, 82.6, 77.6, 48.0, 35.0, 32.8, 27.3; m/z (C.I) 141 (MH+, 90%), 130 ( 61%), 132 (100%), 112 (21%), 99 (11%) C7H10O3 requires 141.0809, found, 141.0549.
4. Conclusions
- In this paper we have shown an expeditious approach to the synthesis of bicylcic lactone using stereocontrol bulky reagents such as K selectride and L-Selectride. These two reagents proved successful in deriving the readily desired syn-alcohol selectivities which cyclises to furnish the desired lactones in average good yields. We have also shown the versatility of Wilkinson’s catalyst in a one-pot synthesis of lactones