-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2012; 2(4): 87-96
doi:10.5923/j.ajoc.20120204.03

Synthesis, Cytotoxicity Evaluation, DFT Molecular Modeling Studies and Quantitative Structure Activity Relationship of Novel 1,8-Naphthyridines
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAhmed A. Fadda , Ahmed M. El Defrawy , Sherihan A. El-Hadidy
Department of Chemistry, Faculty of Science, Mansoura University, El-Gomhoria Street, Mansoura, 35516, Egypt
Correspondence to: Ahmed A. Fadda , Department of Chemistry, Faculty of Science, Mansoura University, El-Gomhoria Street, Mansoura, 35516, Egypt.
| Email: |  |
Copyright © 2012 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

A series of substituted 1,8-naphthyridine derivatives were synthesized from 4-oxo-1,4-dihydro-1,8- naphthyridine-3-carbohydrazide (4) as starting material and investigated as cytotoxic and antitumor agents. Compound 4 reacted with different acid anhydrides to afford the corresponding imides and bis-imide derivatives 5-11. Pyridine and 1,8-naphthyridine derivatives 2-11 were tested for their in vitro cytotoxicity against four different cancer cell lines and structure activity relationship’s (SAR’s) was studied. Compounds 4-11 were studied theoretically using the density functional theory (DFT) with B3LYP/6-31(d) level of calculations, and the electronic properties of these compounds were related to their biological activity.
Keywords: 2-Aminopyridine, Diethylethoxymethylenemalonate, 1,8-Naphthyridine, Acid Anhydrides, Cytotoxicity, Molecular Modeling
Cite this paper: Ahmed A. Fadda , Ahmed M. El Defrawy , Sherihan A. El-Hadidy , Synthesis, Cytotoxicity Evaluation, DFT Molecular Modeling Studies and Quantitative Structure Activity Relationship of Novel 1,8-Naphthyridines, American Journal of Organic Chemistry, Vol. 2 No. 4, 2012, pp. 87-96. doi: 10.5923/j.ajoc.20120204.03.
Article Outline
1. Introduction
- Studies on the synthesis of 1,8-naphthyridines have served as a fertile field of research in the perusal for antibacterial agents[1-3]. Nalidixic acid (1-ethyl-3-carboxy-7- methyl-1,8-naphthyridine-4-one) has been found to be effective particularly against gram negative bacteria found in chronic urinary tract infections[4]. Cancer, a disease of worldwide importance, according to the American cancer society, is a group of disease characterized by uncontrolled growth and spread of abnormal cells. Recently, quinolines and 1,8-naphthyridine are being exploited in cancer chemotherapy and one of the molecules SNS-595 is in second phase of clinical trials[5, 6]. Mammalian topoisomerase ΙΙ is one of the known targets for antitumor agents like doxorubicin, etoposide, ellipticine and amsacrine[7]. 1,8- Nphthyridine derivatives were found to display moderate cytotoxic activity against murine p388 leukemia, when changes were carried out at N-1 and N-7 positions[8, 9]. It has been reported that C-3 carboxamide derivatives with a spacer, which have shown good cytotoxicity along with anti-inflammatory activity[10]. Moreover, pyrroles are important analgesic and anti-inflammatory agents[11, 12]. In view of these facts, and in continuation of our interest in the chemistry of 1,8-naphthyridines[13-17], we undertookthe synthesis and cytotoxicity effects against different carcinoma cells of some new pyrroles containing 1,8- naphthyridine moiety. Protein kinase CK2, formerly known as casein kinase 2, is a ubiquitous and highly conserved serine /threonine kinase that exists in all types of eukaryotic cells. CK2 is a heterotetrameric enzyme, α2β2, αα'β2 or α'2β2, composed of two catalytic subunits (CK2α or CK2α') and two non-catalytic subunits (CK2β)[18]. It plays key roles on cell growth, proliferation, and survival, but over expression of its catalytic subunit can cause cancer[18]. Hence, it can serve as an oncogene in lymphocytes. CK2 is mainly localized in the cytoplasm and the nucleus of normal cells, but it is highly abundant in the nucleus compartment of cancer cells[19]. CK2 was reported to be frequently up regulated in a broad variety of human cancer studies[20-22]. It was shown that over expression of CK2 catalytic subunit results in mammary tumorigenesis and increases a cell's oncogenic potential by sensitizing a cell transformation by other oncogenic proteins[23-26]. It was also shown that the down regulation of CK2 activity decreases cellular proliferation and an increase in the level of apoptosis was observed in cancer cells[27]. In this respect, it was demonstrated that the use of small molecule inhibitors and antisense CK2α resulted in cancer cells death in vitro owing to induction of apoptosis and complete elimination of the tumor was observed with higher concentration of the antitumor[28]. Hence, inhibition of CK2 over expansion has been considered as a viable approach for cancer therapy. With this in mind and with ingoing efforts in our laboratory, using a molecular design approach, this paper reports a variety of synthetic routs leading to the synthesis of some novel 1,8-naphthyridine-3-carboxamide analogues bearing different functional groups, some of which resemble that 5-fluorouracil (5-Fu). Novel analogs were evaluated in vitro against HepG2 (human hepatocellular liver carcinoma cell lines), WI-38 (skin carcinoma cell lines), VERO (cell line was initiated from the kidney of a normal adult African green monkey), and MCF-7 (breast cancer cell lines) and the in vitro cytotoxicity data were correlated with different molecular modeling, predicted for each analogue, to determine the key molecular modeling associated with their biological activity. Quantitative structure activity relationship (QSAR) studies have been widely employed in the pharmaceuticals industry since the 1980’s often by utilizing steric, electrostatic, lipophilicity[29], thermodynamic and electronic properties to correlate molecular structure with biological activity. In this study, DFT methods were used to predict equilibrium molecular geometry of compounds 4-11 including B3LYP energy functional with basis set 6-31G(d).
2. Results and Discussion
2.1. Chemistry
- In view of these observations and in continuation of our previous work in quinoline chemistry[30, 31], we synthesized some new heterocyclic compounds containing quinoline and tested their biological activities. The synthetic procedures adopted to obtain the target compounds are depicted in Schemes 1 and 2. The starting material 2-aminopyridine (1) was refluxed with diethyl 2-(ethoxymethylene)malonate in absolute ethanol to give diethyl 2-((pyridin-2-ylamino) methylene)malonate (2) in high yield. The IR spectrum of 2 showed the presence of absorption band at 1679 cm-1 for ester carbonyl group, and absorption frequency at 3469 cm-1 corresponding to NH function. The mass spectrum of 2 showed the molecular ion peak at m/z 264 (M+) corresponding to the molecular formula (C13H16N2O4). Cyclization reaction of compound 2 was performed by its boiling in diphenyl ether to give ethyl-4-oxo-1,4-dihydro-1,8- naphthyridine-3-carboxylate (3) in good yield. The IR spectrum of 3 showed the presence of characteristic absorption bands at ύ 3411, 1710, 1627, and 1557 cm-1 corresponding to NH, C=O of ester, α,β-unsaturated ketone and C=C functions, respectively. Its 1H-NMR spectrum reveals a triplet signal at δ 1.30 ppm (CH3), a quartet signal at δ 4.31 ppm (CH2), a D2O exchangeable NH at δ 9.17 ppm as singlet signal, and a singlet signal for CH at δ 7.26 ppm besides the aromatic protons of pyridine ring at δ 7.69-8.94 ppm. Moreover, the mass spectrum gave an additional evidence for the structure 3 which showed its molecular ion peak at m/z 218 corresponding to a molecular formula (C11H10N2O3). Furthermore, treatment of β-keto ester 3 with hydrazine hydrate gave the corresponding acid hydrazide 4 in 70% yield. Its structure was proved by means of both elemental analyses and spectral data. The IR spectrum showed the presence of characteristic absorption bands at ύ 3435, 3306, 1685 and 1624 cm-1 corresponding to NH2, NH, amidic C=O, and α,β-unsaturated C=O functions, respectively. Moreover, the mass spectroscopic measurements of 4 gave a more conformation for its correct structure which showed the molecular ion peak at m/z 204 (M+) (Scheme 1).Condensation of starting material 4 with acid anhydrides, namely, phthalic anhydride, 4-nitrophthalic anhydride and 3-nitrophthalic anhydride in refluxing ethanol containing few drops of acetic acid afforded the corresponding N-(1,3-dioxoisoindolin-2-yl)-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxamide (5), N-(5-nitro-1,3-dioxoisoindolin- 2-yl)-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carbox-amide (6) and N-(4-nitro-1,3-dioxoisoindolin-2-yl)-4-oxo-1,4- dihydro-1,8-naphthyridine-3-carboxamide (7), respectively. The IR spectra of compounds 5-7, in general, showed the absence of absorption frequency of NH2 function at 3435 cm-1 of compound 4, and in stead, the appearance of new bands within ύ = 1731-1697 cm-1 corresponding to carboximide groups. The mass spectrum of compound 5 showed the molecular ion peak at m/z 334 (M+). Similarly, the mass spectra of compounds 6 and 7 showed the molecular ion peaks at m/z 379 (M+) corresponding to molecular formula (C17H9N5O6). Similarly, condensation of compound 4 with 3,4,5,6-tetra- bromophthalic anhydride, and 1,2,4-benzenetricarboxylic anhydride (trimellitic anhydride) in refluxing ethanol containing few drops of acetic acid afforded the corresponding N-(4,5,6,7-tetrabromo-1,3-dioxoisoindolin-2-yl)-4-oxo-1,4- dihydro-1,8-naphthyridine-3-carboxamide (8) and 1,3-di- oxo-2-(4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxamide) isoindoline-5-carboxylic acid (9).
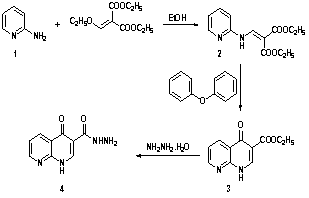 | Scheme 1. Synthesis of 4-oxo-1,4-dihydro-1,8-naphthyridine-3- carbohydrazide (4) |
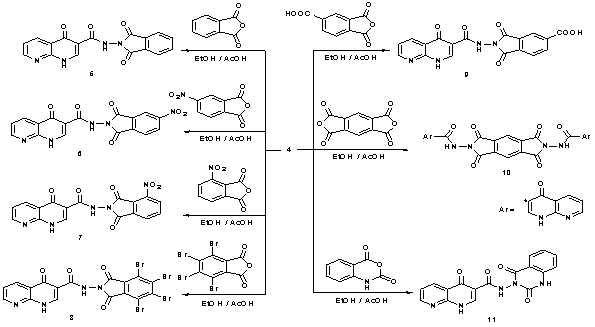 | Scheme 2. Synthesis of 1,8-naphthyridine-3-carboxamide derivatives 5-11 |
 | Figure 1. Routes for the mechanism of formation of 1,8-naphthyridine-3-carboxamide derivatives 5-11 |
2.2. Pharmacology
2.2.1. Cytotoxicity
- Cytotoxicity was expressed as the concentration that caused 50% loss of the cell monolayer (IC50). The assay was used to examine the newly synthesized compounds. 5-Fluorouracil as a standard anticancer drug was used for comparison[32-34]. The results of our preliminary screening indicated that compounds 7, 8, and 10 showed moderate cytotoxicity activity. The other compounds showed weak cytotoxicity activity (Table 1).Subsequently, we may conclude the following structure activity relationship’s (SAR’s). 1) The presence of basic skeleton (phthalimide moiety) is necessary for the broad spectrum of cytotoxic activity towards different cell lines (HepG2, WI-38, VERO and MCF-7) (Fig. 2). 2) Introducing a nitro group (electron withdrawing group, -ve inductive effect) in position 3 of benzene ring in phthalimide moiety increases the activity towards WI-38 and VERO also diminishes the activity towards HepG2 and MCF-7 (compound 7). 3) Introducing four bromine atoms (electron withdrawing group, -ve inductive effect) in benzene ring of phthalimide moiety increases the activity against all cell lines (compound 8). 4) According to the above findings the presence of two phthalimide moieties enhanced the cytotoxicity activity towards all cell lines (compound 10). 5) In compound 11 the presence of NH group near to carbonyl group in isatoic acid moiety acts as electron withdrawing group and showed nearly moderate activity against HepG2 and WI-38. 6) The presence of electron withdrawing group (NO2 and COOH) in position 4 of benzene ring in phthalimide moiety decreases the negative inductive effect on the reaction center of molecule and consequently decreases the cytotoxicity activity (compounds 6 and 9). 7) Compound 5 has phthalimide moiety with no substituents in benzene ring, showed weak activity. 8) Compounds 2, 3 and 4 have no phthalimide moiety, showed weak cytotoxicity activity.
|
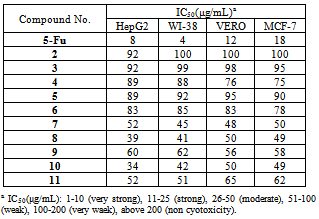
 | Figure 2. Confluent Monolayers of cell lines used for testing |
2.3. Molecular Modeling
- DFT methods were used to predict equilibrium molecular geometry of compounds 4-11 including (B3LYP) energy functional with basis set 6-31G (d). Some electronic properties, such as HOMO and LUMO energy values, frontier orbital energy gap, molecular dipole moment (μ), and molecular electrostatic potential (MEP) were calculated and have been used to understand the properties and activity of the newly prepared compounds.
2.3.1. Electronic Properties
- The frontier orbitals, HOMO and LUMO determine the way a molecule interacts with other species. The HOMO is the orbital that primarily acts as an electron donor and the LUMO is the orbital that largely acts as the electron acceptor. The frontier orbital gap helps characterize the chemical reactivity and kinetic stability of the molecule. A molecule with a small frontier orbital gap is more polarizable and is generally associated with a high chemical reactivity, low kinetic stability and is also termed as soft molecule[35].The 3D plots of the frontier orbitals HOMO, LUMO and the molecular electrostatic potential map (MESP) for the molecules are shown in Fig. 3 and Fig. 4, respectively. It can be seen from Fig. 3 that, the HOMO is almost distributed uniformly in all case on the 1,8-naphthyridine moiety part of the molecules, HOMO’s of the molecules show considerable π bond character. The LUMO in all case is found to be mainly phthalimide moiety part of the molecules except for the LUMO of the compound 11 compound which is located on the 1,8-naphthyridine moiety part of the molecule. Differently, HOMO and LUMO energy values seem to be related to the different reactivity for these compounds, consequently different biological activity profile (Table 1). The frontier orbital gap in case of compounds 7 and 8 is found to be 3.62545 eV and 3.51334 eV respectively in case of using DMSO as a solvent (which showed better biological activities) are considered as a small values compared to the frontier orbital gap for the other compounds except for the compound 6. Compound 11 shows a moderate biological activity (Table 1), in spite of showing a higher frontier orbital gap. Although there is a considerable variation between the frontier orbital gaps for the compounds under investigation, the close values of this physicochemical property don't allow the identification of which compound is the most reactive one.
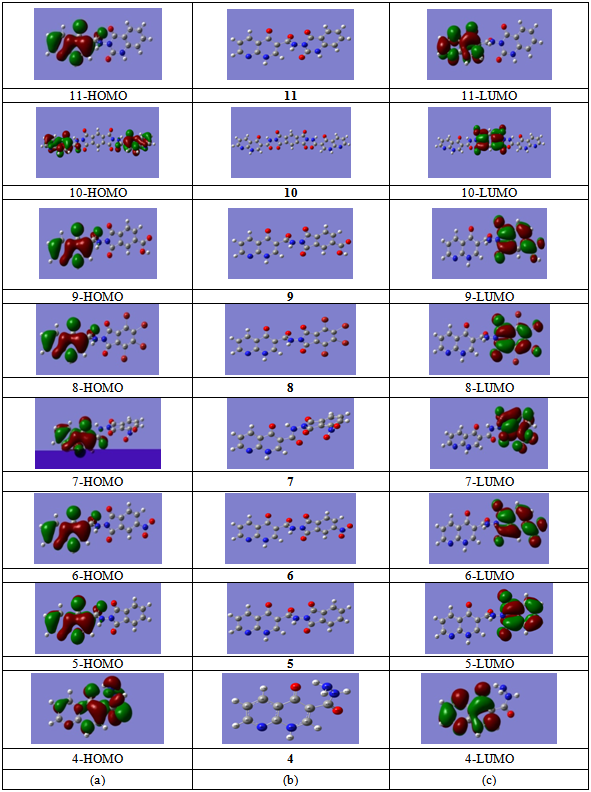 | Figure 3. (a) HOMO plots of compounds at B3LYP/6-31G (d). (b) Optimized structures of compounds at B3LYP/6-31G (d). (c) LUMO plots of compounds at B3LYP/6-31G (d) |
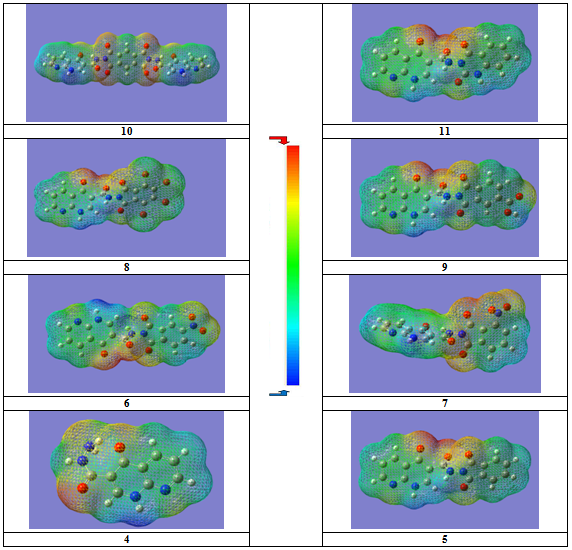 | Figure 4. MESP of different compounds under investigation |
2.3.2. Electric Moments
- The calculated dipole moment for all the molecules is given in Table 2. The dipole moment in a molecule is an important property that is mainly used to study the intermolecular interactions involving the non bonded type dipole-dipole interactions, because higher the dipole moment, stronger will be the intermolecular interactions, which agrees with the experimental results.
3. Conclusions
- In summary modification of ethyl-4-oxo-1,4- dihydro-1,8-naphthyridine-3-carboxylate (3) and 4-oxo-1,4- dihydro-1,8-naphthyridine-3-carbohydrazide (4) derivatives produced compounds with potential with further development as anticancer agents. Based on these preliminary screening results, compounds 7, 8 and 10 showed significant activity in certain cancer cell and have been targeted for further studies. Compounds 8 and 10 showed the highest cytotoxic activity. Meanwhile, compounds 2, 3 and 4 proved to have the weakest activity. Different (QSAR) models using the MLR techniques were developed and showed reasonable agreement with the experimental data. It was shown that the best five molecular descriptors were dipole moment, excitation energy, energy of the LUMO, solvent accessible surface area, and total heat of formation. Recently, the cancer chemopreventive effects of 1,8-naphthyridino-phthalimide derivatives have been intensively investigated. Studies are underway in our laboratory to investigate the more derivatives of such model of compounds toward different carcinoma line cells.
4. Experimental
4.1. Instruments
- All melting points are recorded on Gallenkamp electric melting point apparatus and are uncorrected. The IR spectra υ cm-1 (KBr) were recorded on Perkin Elmer Infrared Spectrophotometer Model 157, Grating. The 13C- and 1H-NMR spectra were run on Varian Spectrophotometer at 400 MHz and 100 MHz. using TMS as an internal reference and DMSO-d6 as solvent. The mass spectra (EI) were recorded on 70 eV with Kratos MS equipment and/or a Varian MAT 311 A Spectrometer at Cairo Univ., Giza, Egypt, and at Assiut university central lab. Elemental analyses (C, H, and N) were carried out at the Microanalytical Center of Cairo University, Giza, Egypt. The results were found to be in good agreement with the calculated values.
4.1.1. Synthesis of Diethyl 2-((Pyridin-2-Ylamino) Methylene)Malonate (2)
- To a solution of 2-aminopyridine (1) (0.94 g, 0.01 mmol) in absolute ethanol (25 mL) and diethyl 2-(ethoxy methylene)malonate (2.16 g, 0.01 mmol) was added. The reaction mixture was refluxed for 4 h and left to cool at room temperature. The precipitated solid product was filtered off, dried and recrystallized from ethanol to give compound 2. Yield (98%); white crystals; m.p. 64℃; IR (KBr): ν/cm-1= 3469 (NH), 1679, 1650 (2 CO, ester); 1H-NMR(DMSO-d6) δ (ppm): 1.28 (t, 6H, 2 x CH3), 4.30 (q, 4H, 2 x CH2), 7.32 (s, 1H, CH), 6.68-8.02 (m, 4H, Ar-H), 11.81 (s, 1H, NH); 13C-NMR(DMSO-d6) δ (ppm): 171.3, 170.9, 162.7, 156.3, 150.1, 141.6, 114.7, 112.2, 101.8, 67.8, 67.7, 15.3, 15.1; MS (EI, 70 ev) m/z (%) = 264 (M+, 97.86), 219 (97.19), 191 (99.94), 173 (89.35), 163 (100), 146 (69.12). Anal. Calcd. for C13H16N2O4 (264.28): C, 59.08; H, 6.10; N, 10.60%. Found: C, 59.15; H, 6.03; N, 10.54%.
4.1.2. Synthesis of Ethyl-4-Oxo-1,4-Dihydro- 1,8- Naphthyridine-3-Carboxylate (3)
- The cyclization reaction was performed by adding portions of 2 (2.64g, 0.01 mmol) to boiling diphenyl ether. The reaction mixture was refluxed for 1 h, cooled to room temperature, then added petroleum ether. The cooled precipitation was washed with diethyl ether and dried. The resulting product recrystallized from ethanol to give compound 3. Yield (85%); pale brown powder; m.p. 72℃; IR (KBr): ν/cm-1= 3411 (NH), 1710 (CO, ester), 1627 (α,β-unsaturated C=O), 1557 (C=C); 1H-NMR(DMSO-d6) δ (ppm): 1.30 (t, 3H, CH3), 4.31 (q, 2H, CH2), 7.26 (s, 1H, CH), 7.69 (m, 1H, β-CH of pyridine ring), 7.85 (m, 1H, γ-CH of pyridine ring), 8.94 (t, 1H, α-CH of pyridine ring), 9.17 (s, 1H, NH); 13C-NMR(DMSO-d6) δ (ppm): 178.9, 171.5, 154.3, 147.7, 150.0, 140.3, 125.3, 114.9, 113.2, 67.9, 14.8; MS (EI, 70 ev) m/z (%) = 218 (M+, 32.24), 173 (64.54), 146 (100), 118 (58.92), 78 (79.68). Anal. Calcd. for C11H10N2O3 (218.21): C, 60.55; H, 4.62; N, 12.84%. Found: C, 60.62; H, 4.60; N, 12.97%.
4.1.3. Synthesis of4-Oxo-1,4-Dihydro-1,8-Naphthyridine-3- Carbohydrazide (4)
- To a solution of 3 (2.18 g, 0.01 mmol) in ethanol (50 mL) and hydrazine hydrate (1.5 g, 0.01 mmol) was added. The reaction mixture was refluxed for 6 h then cooled to room temperature. The precipitated solid was filtered off, dried, and recrystallized from ethanol to give compound 4. Yield (70%); white crystals; m.p. 218o℃; IR (KBr): ν/cm-1= 3435 (NH2), 3306 (NH), 1685 (C=O, amidic), 1624 (α,β- unsaturated C=O); 1H-NMR(DMSO-d6) δ (ppm): 4.43 (s, 2H, NH2), 7.25 (s, 1H, CH), 7.71-8.97 (m, 3H, Ar-H), 9.28 (s, 1H, NH), 10.8 (s, 1H, NH); 13C-NMR(DMSO-d6) δ (ppm): 179.1, 172.4, 154.2, 152.6, 147.3, 140.3, 125.4, 115.1, 114.7; MS (EI, 70 ev) m/z (%) = 205 (M++1, 0.5), 204 (M+, 37.6), 172 (100), 99 (40.4), 74 (69.8). Anal. Calcd. for C9H8N4O2 (204.19): C, 52.94; H, 3.95; N, 27.44%. Found: C, 52.88; H, 3.81; N, 27.69%.
4.1.4. General procedure for the syntheses of 1,8- naphthyridine-3-carboxamide derivatives 5-10
- A mixture of compound 4 (0.01 mmol) and anhydride derivatives namely; phthalic anhydride (0.01 mmol), 4-nitrophthalic anhydride (0.01 mmol), 3-nitrophthalic anhydride (0.01 mmol), 3,4,5,6-tetrabromophthalic anhydride (0.01 mmol), 1,2,4-benzenetricarboxylic anhydride (trimellitic anhydride) (0.01 mmol) or 1,2,4,5-benzene- tetracarboxylic dianhydride (0.01 mmol) in ethanol (15 mL) containing glacial acetic acid (5 drops) were refluxed for 2 h. The reaction mixture was left to cool at room temperature; the precipitated solid product was filtered off, dried and recrystallized from ethanol to give compounds 5-10, respectively.
4.1.4.1. N-(1,3-Dioxoisoindolin-2-Yl)-4-Oxo-1,4-Dihydro- 1,8-Naphthyridine-3-Carboxamide (5)
- Yield (90%); pale yellow powder; m.p. sharing at 330oC; IR (KBr): ν/cm-1= 3355 (NH), 1683 (CO, imide), 1631 (C=O, amidic), 1612 (α,β-unsaturated C=O), 1579 (C=C); 1H-NMR(DMSO-d6) δ (ppm): 7.29 (s, 1H, CH), 7.70-8.92 (m, 7H, Ar-H), 9.33 (s, 1H, NH), 10.6 (s, 1H, NH); 13C-NMR(DMSO-d6) δ (ppm): 178.7, 172.6, 172.5, 172.4, 154.3, 152.8, 146.6, 140.1, 133.4, 133.3, 133.2, 133.0, 129.1, 129.0, 125.3, 115.2, 114.1; MS (EI, 70 ev) m/z (%) = 335 (M++1, 4.8), 334 (M+, 17.3), 204 (26.2), 172 (100), 147 (7.2), 145 (2.4), 118 (0.9), 103 (27.5), 76 (24.8). Anal. Calcd. for C17H10N4O4 (334.29): C, 61.08; H, 3.02; N, 16.76%. Found: C, 61.29; H, 3.30; N, 16.58%.
4.1.4.2. N-(5-Nitro-1,3-Dioxoisoindolin-2-Yl)-4-Oxo-1,4- Dihydro- 1,8-Naphthyr-Idine-3-Carboxamide (6)
- Yield (82%); white powder; m.p. 331℃; IR (KBr): ν/cm-1= 3448 (NH), 1731-1697 (CO, imide), 1625 (α,β-unsaturated C=O), 1334, 1538 (NO2); 1H-NMR (DMSO-d6) δ (ppm): 7.21 (s, 1H, CH), 7.67-8.83 (m, 6H, Ar-H), 9.21 (s, 1H, NH), 10.1 (s, 1H, NH); 13C-NMR (DMSO-d6) δ (ppm): 178.6, 172.5, 172.4, 172.3, 154.1, 153.6, 152.7, 146.7, 140.1, 133.4, 129.3, 128.6, 125.31, 123.4, 115.6, 114.7; MS (EI, 70 ev) m/z (%) = 379 (M+, 6.0), 173 (100), 145 (3.0), 99 (28.0), 74 (45.0). Anal. Calcd. for C17H9N5O6 (379.28): C, 53.83; H, 2.39; N, 18.46%. Found: C, 53.98; H, 2.37; N, 18.34%.
4.1.4.3. N-(4-nitro-1,3-dioxoisoindolin-2-yl)-4-oxo-1,4- dihydro-1,8-naphthyr-idine-3-carboxamide (7)
- Yield (89%); pale yellow powder; m.p. 135℃; IR (KBr): ν/cm-1= 3445 (NH), 1734-1690 (CO, imide), 1627 (α,β-unsaturated C=O); 1H-NMR(DMSO-d6) δ (ppm): 7.23 (s, 1H, CH), 7.61-8.79 (m, 6H, Ar-H), 9.23 (s, 1H, NH), 10.4 (s, 1H, NH); 13C-NMR(DMSO-d6) δ (ppm): 178.5, 173.0, 172.6, 172.3, 154.1, 152.7, 151.1, 146.7, 140.0, 134.1, 133.7, 133.2, 129.1, 129.0, 125.3, 115.8, 114.9; MS (EI, 70 ev) m/z (%) = 379 (M+, 5.2), 269 (30.7), 173 (100), 146 (9.3), 99 (26.1), 74 (29.3). Anal. Calcd. for C17H9N5O6 (379.28): C, 53.83; H, 2.39; N, 18.46%. Found: C, 53.97; H, 2.33; N, 18.41%.
4.1.4.4. N-(4,5,6,7-Tetrabromo-1,3-Dioxoisoindolin-2-Yl)- 4-Oxo-1,4-Dihydro-1,8-Naphthyr-Idine-3-Carboxamide (8)
- Yield (96%); white powder; m.p. 353℃; IR (KBr): ν/cm-1= 3234 (NH), 1739, 1704 (CO, imide), 1660 (C=O, amidic), 1627 (α,β-unsaturated C=O); 1H-NMR(DMSO-d6) δ (ppm): 7.33 (s, 1H, CH), 7.73-8.85 (m, 3H, Ar-H), 9.35 (s, 1H, NH), 11.1 (s, 1H, NH); 13C-NMR(DMSO-d6) δ (ppm): 178.1, 172.6, 172.3, 172.1, 154.0, 152.7, 146.4, 141.2, 140.9, 140.2, 130.1, 130.0, 125.8, 125.6, 125.1, 115.3, 114.6; MS (EI, 70 ev) m/z (%) = 651 (M++1, 0.7), 650 (M+, 0.8), 475 (1.2), 466 (0.7), 392 (0.9), 173 (100), 145 (5.5), 105 (25.1), 85 (1.1), 77 (40.8). Anal. Calcd. for C17H6N4O4Br4 (649.87): C, 31.42; H, 0.93; N, 8.62%. Found: C, 31.65; H, 0.76; N, 8.64%.
4.1.4.5. 1,3-Dioxo-2-(4-Oxo-1,4-Dihydro-1,8-Naphthyridine- 3-Carboxamide)Iso-Indoline-5-Carboxylic Acid (9)
- Yield (90%); yellow powder; m.p. 324℃; IR (KBr): ν/cm-1= 3449 (NH), 1700 (CO, imide), 1627 (α,β- unsaturated C=O); 1H-NMR(DMSO-d6) δ (ppm): 7.35 (s, 1H, CH), 7.71-8.97 (m, 6H, Ar-H), 9.32 (s, 1H, NH), 10.9 (s, 1H, NH), 13.7 (s, 1H, OH); 13C-NMR(DMSO-d6) δ (ppm): 178.1, 173.4, 172.5, 171.8, 171.6, 154.1, 152.6, 146.4, 140.2, 138.6, 134.2, 134.0, 132.3, 128.1, 127.9, 125.1, 115.7, 114.8; MS (EI, 70 ev) m/z (%) = 378 (M+, 0.1), 204 (35.7), 190 (0.5), 173 (100), 99 (32.2), 74 (41.3). Anal. Calcd. for C18H10N4O6 (378.3): C, 57.15; H, 2.66; N, 14.81%. Found: C, 57.15; H, 2.87; N, 14.90%.
4.1.4.6. N,N'-(1,3,5,7-Tetraoxopyrrolo[3,4-F]Isoindole- 2,6- (1H,3H,5H,7H)-Diyl)Bis(4-Oxo-1,4-Dihydro-1,8- Naphthyridine- 3-Carboxamide) (10)
- Yield (94%); light yellow powder; m.p. sharing at 340℃; IR (KBr): ν/cm-1= 3450 (NH), 1689 (CO, imide), 1629 (α,β-unsaturated C=O); 1H-NMR(DMSO-d6) δ (ppm): 7.29 (s, 2H, 2 x CH), 7.75-8.84 (m, 8H, Ar-H), 9.31 (s, 2H, 2 x NH), 10.6 (s, 2H, 2 x NH); 13C-NMR(DMSO-d6) δ (ppm): 178.1, 177.8, 172.1, 172.0, 171.8, 171.6, 171.4, 171.1, 154.3, 154.1, 152.3, 152.0, 146.0, 145.8, 140.1, 139.7, 135.2, 135.1, 135.0, 125.1, 125.0, 124.9, 115.4, 115.0, 114.7, 114.3; MS (EI, 70 ev) m/z (%) = 590 (M+, 0.7), 562 (M+_ CO, 1.9), 246 (17.6), 173 (100), 145 (3.2), 117 (0.7), 99 (21.8), 74 (22.7). Anal. Calcd. for C28H14N8O8 (590.46): C, 56.96; H, 2.39; N, 18.98%. Found: C, 56.76; H, 2.54; N, 18.97%.
4.1.5. N-(1,2-Dihydro-2,4-Dioxoquinazolin-3(4H)-Yl)-4- Oxo-1,4-Dihydro-1,8-Naphthyri-Dine-3-Carboxamide (11)
- A mixture of compound 4 (2.04g, 0.01 mmol) and isatoic anhydride (1.63 g, 0.01 mmol) in ethanol and glacial acetic acid (50 mL) were stirred for 3 h at room temperature. The resulting crystals were filtered off, dried and recrystallized from ethanol to give compound 11. Yield (82%); yellow crystals; m.p. 247℃; IR (KBr): ν/cm-1= 3430 (NH), 1677 (CO, imide), 1619 (α,β-unsaturated C=O); 1H-NMR (DMSO-d6) δ (ppm): 7.31 (s, 1H, CH), 6.89-8.76 (m, 7H, Ar-H), 9.35 (s, 1H, NH), 11.1 (s, 1H, NH); 13C-NMR(DMSO-d6) δ (ppm): 178.0, 172.5, 171.6, 171.4, 154.1, 152.5, 146.4, 140.3, 138.3, 132.7, 132.5, 127.8, 125.3, 125.1, 122.3, 115.7, 114.9; MS (EI, 70 ev) m/z (%) = 349 (M+, 0.4), 219 (1.4), 206 (1.1), 202 (0.3), 173 (100), 147 (0.8), 145 (3.1), 133 (89.7), 120 (49.6), 99 (37.1). Anal. Calcd. for C17H11N5O4 (349.3): C, 58.45; H, 3.17; N, 20.05%. Found: C, 58.75; H, 3.05; N, 20.21%.
4.2. Cytotoxicity Activity[35-37]
- RPMI-1640 medium (Sigma Co., St. Louis, USA), Foetal Bovine serum (GIBCO, UK), and the cell lines from ATCC were used.The cytotoxic activity of the synthesized compounds was tested against human hepatocellular liver carcinoma cell line (HepG2), human lung fibroblast cell line (WI-38), human causcasian breast adenocarcinoma cell line (MCF-7), and normal adult African green monkey kidney cell line (VERO). The stock samples of the compounds were diluted with RPMI-1640 medium to desired concentrations ranging from 10 to 1000 μg/mL. The final concentration of DMSO in each sample did not exceed 1% v/v.The cells were bath-cultured for 10 d, then seeded in 96 well plates of 10×103 cells/well in fresh complete growth medium in 96-well microtiter plastic plates at 37℃ for 24 h under 5% CO2 using a water jacketed carbon dioxide incubator (Shedon. TC2323. Cornelius, OR, USA). The medium (without serum) was added and cells were incubated either alone (negative control) or with different concentrations of sample to give a final concentrations of (1000, 500, 200, 100, 50, 20, 10 μg/mL). Cells were suspended in RPMI-1640 medium, 1% antibiotic-antimycotic mixture (104 u/mL) potassium penicillin, 104 μg/mL streptomycin sulfate and 25 μg/mL Amphotericin B ) and 1% L-glutamin in 96-well flat bottom microplates at 37℃ under 5% CO2. After 96 h of incubation, the medium was again aspirated, trays were inverted onto a pad of paper towels, the remaining cells rinsed carefully with medium, and fixed with 3.7% (v/v) formaldehyde in saline for at least 20 min. The fixed cells were rinsed with water. The %viability of cells was examined visually as described previously[42].
4.3. Computational Details
- Gaussian 03[43], software package was used for theoretical calculation. The quantum chemical calculations were performed applying DFT method, with Beeke-3-Lee- Yang-Parr (B3LYP) supplemented with the standard 6-31G(d) basis set. All compounds have been optimized, where the ground state geometries and the frontier molecular orbital characteristics were analyzed on the optimized structures at the same level. In all cases, the steady state nature (minimum on the potential energy surface) of the optimized compounds has been confirmed by calculating the corresponding frequencies at the same computational level. The optimized geometry of the compounds under investigation in their ground states has been performed with the aim perform structure-activity relationship’s (SAR’s) studies, some electronic properties, such as HOMO and LUMO energy values, frontier orbital energy gap, molecular dipole moment (µ), and molecular electrostatic potential (MEP) were calculated and have been used to understand the properties and activity of the newly prepared compounds. MEP isoenergy surface maps were generated in the range from -50.0 (deepest red color) to +50.0 (deepest blue color) kcal mol-1. Based on the optimized gas phase geometry, single point solvation calculations were conducted using the isoelectric focusing polarized continuum model (IEF-PCM) [44-46], at the B3LYP/6-31G(d) level of theory. What’s more, the dielectric constant of DMSO (εDMSO = 46.7) was used to approximate the bulk effects of solvation.
ACKNOWLEDGEMENTS
- To Prof. Dr. Farid A. Badria, Pharmacognosy Department, Faculty of Pharmacy, Mansoura University, for microbiological screening, he is greatly acknowledged.
References
| [1] | H. Egawa, T. Miyamota, A. Minamida, Y. Nishimura, H. Okada, H. Uno, T. Motosumota, J. Med. Chem. 27 (1984) 1543-1548. |
| [2] | K. Kohima, M. Motoyoshi, Japan Kotai Tokyo JP (1988) 01,100,603; Chem. Abstr. 109 (1988) 189591. |
| [3] | C.S. Cooper, P.L. Klock, D.T.W. Chu, D.J. Hardy, R.N. Swanson, J.J. Plattner, J. Med. Chem. 35 (1992) 1392-1398. |
| [4] | J. Nezval, K. Halocka, Experientia 23 (1967) 1043-1044. |
| [5] | C.T. Supuran, A. Scozzafava, Expert Opin. Ther. Patent 14 (2004) 35-53. |
| [6] | FDA News Drug Pipeline Alert, March 21, vol. 4 (2006) No. 56. |
| [7] | D.A. Burden, N. Oshereroff, Biochim. Biophys. Acta, 1400 (1998) 139-154. |
| [8] | K. Tomita, Y. Tsuzuki, K. Shibamori, M. Tshima, F. Kajikawa, Y. Sato, S. Kashimoto, K. Chiba, K. Hino, J. Med. Chem. 45 (2002) 5564-5575. |
| [9] | Y. Tsuzuki, K. Tomita, K. Shibamori, Y. Sato, S. Kashimoto, K. Chiba, J. Med. Chem. 47 (2004) 2097-2109. |
| [10] | S.K. Srivastava, M. Jaggi, A.T. Singh, A. Madaan, N. Rani, M. Vishnoi, S.K. Agarwal, R. Mukherjee, A.C. Burman, Bioorg. Med. Chem. Lett. 17 (2007) 6660-6664. |
| [11] | C. D. Lunsford, A. D. Cale Jr., J. W. Ward, B. V. Franko, H. Jenkins, J. Med. Chem. 7 (1964) 302-310. |
| [12] | C.A. Miller, L.M. Long, J. Am. Chem. Soc. 73 (1951) 4895-4898. |
| [13] | V.J. Ram, M. Nath, S. Chandra, Indian J. Chem. 33B (1994) 1048-1052. |
| [14] | M.M. Dutta, B.N. Goswami, J.C.S. Kataky, Indian J. Chem. Soc. 64 (1987) 195-197. |
| [15] | B. Kalluraya, R. Chimbalkar, B. Shivarama Holla, Indian J. Heterocycl. Chem. 5 (1995) 37-40. |
| [16] | H.P. Shah, B.R. Shah, J.J. Bhatt, N.C. Desai, P.B. Trivedi, N.K. Undavia, Indian J. Chem. 37B (1998) 180-182. |
| [17] | K. Mogilaiah, K.R. Reddy. G.R. Rao, B. Sreenivasulu, Coll. Czech. Chem. Commun. 53 (1988) 1539-1542. |
| [18] | (a) Z. Nie, C. Perretta, P. Erickson, S. Margosiak, J. Lu, A. Averill, R. Almassy, S. Chu, Bioorg. Med. Chem. Lett. 18 (2008) 619-623; (b) Cambridge Crystallographic Data Base. |
| [19] | K.A. Ahmed, G. Wang, J. Slaton, G. Unger, K. Ahmed, Anti-Cancer Drugs 16 (2005) 1037-1043. |
| [20] | S. Tawfic, S. Yu, H. Wang, R. Faust, A. Davis, K. Ahmed, Histl. Histopathol. 16 (2001) 573-582. |
| [21] | B. Guerra, O. G. Issinger, Electrophoresis 20 (1999) 391-408. |
| [22] | S. Saha, A. Badelli, P. Buckhaults, V.E. Velculescu, C. Roger, B.S. Croix, K.E. Romans, M.A. Choti, C. Lengauer, K.W. Kinzler, B. Vogelstein, Science 294 (2001) 1343-1346. |
| [23] | D. C. Seldin, P. Leder, Science 267 (1995) 894-897. |
| [24] | M.A. Kelliher, D.C. Seldin, P. Leder, EMBO J. 15 (1996) 5160-5166. |
| [25] | E. Ladesman-Bollag, P.L. Channavajhala, R.D. Cardiff, D.C. Seldin, Oncogene 16 (1998) 2965-2974. |
| [26] | P. Channavajhala, D.C. Seldin, Oncogene, 21 (2002) 5280-5288. |
| [27] | G. Wang, G. Unger, K.A. Ahmed, J.W. Slaton, K. Ahmed, Mol. Cell. Biochem. 274 (2005) 77-84. |
| [28] | J.W. Slaton, G.M. Unger, D.T. Sloper, A.T. Davis, K. Ahmed, Cancer Res. 2 (2004) 668-677. |
| [29] | G.E. Kellog, S.F. Semus, D.J. Abraham, J. Comput-Aided Mol. Des. 5 (1991) 545-552. |
| [30] | A.A. Fadda, M.M. Youssif, H.M. Hassan, Indian J. Chem. 26B (1987) 994-997. |
| [31] | A.A. Fadda, A.M. Khalil, M.M. El-Habbal, Pharmazie 46 (1991) 743-744. |
| [32] | C.K. Lee, H. Jiang, A.M. Scofield, J. Carbohydr. Chem. 16 (1997) 49-62. |
| [33] | H.A. Abdel-Aziza, B.F. Abdel-Wahab, F.A. Badria, Arch. Pharm. 343 (2010) 152-159. |
| [34] | B. Hegazi, H.A. Mohamed, K.M. Dawood, F.A. Badria, Chem. Pharm. Bull. 58 (2010) 479-483. |
| [35] | E. Scrocco, J. Tomasi, Advances in Quantum Chemistry, Academic Press, New York, Vol. 11 (1978) pp. 115-194. |
| [36] | I. Fleming, Frontier Orbitals and Organic Chemical Reactions, John Wiley and Sons, New York, (1976) pp. 5-27. |
| [37] | J.S. Murray, K. Sen, Molecular Electrostatic Potentials, Concepts and Applications, Elsevier, Amsterdam (1996). |
| [38] | I. Alkorta, J.J. Perez, Int. J. Quant. Chem. 57 (1996) 123-135. |
| [39] | F.J. Luque, M. Orozco, P.K. Bhadane, S.R. Gadre, J. Phys. Chem. 97 (1993) 9380-9384. |
| [40] | J. Sponer, P. Hobza, Int. J. Quant. Chem. 57 (1996) 959-970. |
| [41] | A.K. Bhattacharjee, R.K. Gupta, D. Ma, J.M. Karle, J. Mol. Recognit. 13 (2000) 213-220. |
| [42] | T. Fouad, C. Nielsen, L. Brunn, E.B. Pederson, Sci. J. Az. Med. Fac. (Girls) 19 (1998) 1173-1187. |
| [43] | M.J. Frisch, G.W. Trucks, M. Ishida, T. Nakajima, J.B. Foresman, et al., J.A. Pople, Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford CT, (2004). |
| [44] | B. Mennucci, E. Cances, J. Tomasi, J. Phys. Chem. B. 101 (1997) 10506-10517. |
| [45] | B. Mennucci, J. Tomasi, J. Chem. Phys. 106 (1997) 5151-5158. |
| [46] | J. Tomasi, B. Mennucci, E. Cances, J. Mol. Struct. (THEOCHEM) 464 (1999) 211-226. |