-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2012; 2(4): 74-78
doi: 10.5923/j.ajoc.20120204.01
Some Transformative Reactions of Odollactone
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLPranab Ghosh , Utpal Debnath , B. P. Pradhan
Natural Product and Polymer Chemistry Laboratory, Department of Chemistry, University of North Bengal, Darjeeling, West Bengal -734 013, India
Correspondence to: Pranab Ghosh , Natural Product and Polymer Chemistry Laboratory, Department of Chemistry, University of North Bengal, Darjeeling, West Bengal -734 013, India.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Oxidative rearrangement of odollactone has been carried out with hydrogen peroxide containing p-toluene sulphonic acid in dichloromethane. The products obtained were characterised by chemical transformation using selenium dioxide in acetic acid, sodium dichromate in acetic acid and Li in ethylenediamine followed by spectral analysis. The introduction of olefinic double bond at AB-ring juncture influences the cleavage of γ-lactone ring to the diol in contrast to the parent odollactone that gives the carboxylic acid.
Keywords: Wagner Rearrangement, Friedelane Skeleton, Thermodynamically Stable, Heteroannular System, Li in Ethylenediamine
Article Outline
1. Introduction
- Although structure (1) for odollactone[1,2] has been characterized[3-6] from our laboratory from the plant Gynocardia Odorata available in the hill area of Darjeeling, detailed investigation in respect of its chemical activity has not yet been attempted so far. Keeping this view in mind and to explore the chemical activity of this naturally occurring 3β-hydroxy lactone of friedelane skeletone, we have studied some oxidative and reductive transformations on it. During the investigation some interesting observations were noted which comprise the subject matter of this report.
2. Result and Discussion
- It has been reported[7] that friedelanol when heated with p-toluene sulfonic acid, undergoes rearrangement of the friedelane skeleton to oleanane skeleton via Wagner rearrangement[8]. Odollactone has a γ-lactone function attached to the C-13 and C-15 atoms of friedelane skeleton, thus it is anticipated that movement of the carbocation is rather impossible beyond C-14 position as the migration of C-27 carbonyl carbon from C-13 to C-14 position will give rise to the formation of highly strained β-lactone. Therefore, it was assumed that the carbocation generated at C-3 will have a restricted migration up to C-5 position giving rise to olefinic double bonds at C-5(6)/5(10)/1(10) of glutene[9] derivative (2) or it may further migrate up to C-9 forming double bond at C-8(9)/C-9(11)/ or C-7/(8) of multiflurene/baurene[10] derivative (3). Under the strongly acidic condition the formation of thermodynamically more stable tetrasubstituted olefins at C-5(10) or C-8(9) position is more likely (isomer2or 3).
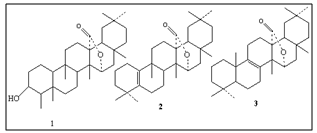 | Figure 1. Structure of odollactone (1), glutene derivative (2), multiflurene/baurene derivative (3) |
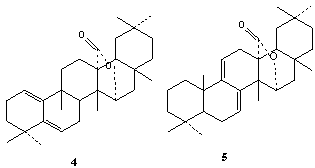 | Figure 2. Structure of glut-1(10),5(6)-dien-27→15α olide (4) and multifluor-7(8),9(11)-diene-27→15α olide (5) |
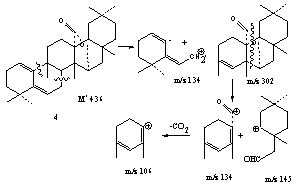 | Scheme 1. Mass fragmentation of compound (4) |
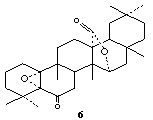 | Figure 3. Structure of glut-5α,10α-epoxy-27→15α-olide (6) |
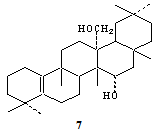 | Figure 4. Li/EDA converts lactone (2) to a diol (7) |
3. Experimental
- Materials and MethodMelting points are uncorrected. Petroleum had b.p. 60-80℃. PMR spectra (chemical shifts in δ, ppm; TMS as internal standard) and 13C NMR spectra were recorded in CDCl3 Bruker avance 300 MHz FT NMR spectrometer, Brucker WH-400 and Brucker WH-270 with DEPT programme respectively; IR spectra in nujol on Beckman IR-20 and Perkin-Elmer FTIR spectrophotometer using KBr discs; UV spectra in methanol on Shimadzu UV-240 spectrometer; mass spectra in Varian MAT 711 instrument at 70 eV. All the chemicals were reagent grade (from Merck, Fluka, SRL) and were purified prior to their use following the standard protocols. Column chromatography was performed over silica gel. TLC was performed over silica gel G (BDH 60-120 mesh).
3.1. Treatment of Compound (1) with H2O2-pTsOH
- To a solution of (1) (2.0 g) in CH2Cl2 (100 mL) was added to a solution 80 mL prepared by mixing p-TsOH (3 g) and 30% H2O2 (5 mL) in tBuOH (80 mL). The mixture was stirred slowly for 24 hrs and poured into water. It was then extracted with CH2Cl2, washed with water, dried by Na2SO4 and the solvent removed under reduced pressure. The residue (1.5 g) was purified by column chromatography over neutral silica gel. Crystallization from CHCl3-MeOH furnished a crystalline solid (A), m.p. 285 ℃. The compound (A) was analyzed for C30H46O2. It responded positive to TNM test indicating the presence of olefinic double bond. IR: 1775 cm-1. MS: m/z at 438 [M+], 423 (base peak) [M+-CH3] and other peaks are m/z 377, 353, 341, 257, 255, 239, 201, 189, 175 and 123. PMR: δ 0.92-1.15 ppm (7 x t-Me protons), 4.32 ppm lactyl proton of C-15 (t, 1H, J=3Hz). 13C NMR: seven t-Me groups at δ 13.7, 19.4, 27.9, 28.7, 29.7, 31.3 and 34.3 ppm as quartets; eleven methylene carbons at δ 19.8, 20.5, 22.2, 24.6, 26.4, 31.7, 34.1, 34.2, 35.9, 39.5, 40.5 ppm as triplet, three doublets at δ 43.7, 44.0 and 80.8 ( C-15 lactyl oxygen) and nine singlets of which six peaks at δ 28.1, 30.4, 33.9, 36.8, 47.8 and 50.9 ppm were due sp3 carbon, at δ 180.3 ppm (lactone carbonyl carbon), at δ 133.3 and 136.1 ppm for double bond.
3.2. SeO2 Oxidation of Compound (2)
- Compound (2) (200 mg) was refluxed in glacial acetic acid (30 mL) with SeO2 (150 mg) for 3 hrs. After usual work up and purification by chromatography and repeated crystallization from CHCl3-MeOH a crystalline solid (A-1) was obtained. The molecular formula of the compound is C30H44O2. m.p: 295-96 ℃, [α] D +75◦. UV: λmax at 233 nm and 237 nm; IR: 1760 cm-1, 820 cm-1. MS: m/z 436 [M+], other fragments at m/z 421, 375, 287, 239, 213, 202, 173, 145, 134 (base). PMR: six singlet for seven tertiary methyl at δ 0.9 (2-CH3), 0.96, 0.97, 1.04, 1.08, 1.18 ppm; δ 4.26 ppm (t, 1H, J=3 Hz) and δ 5.4 and 5.51 ppm as triplet with J = 3 Hz for two olefinic protons. 13C NMR: Seven tertiary methyls appeared as quartet at δ 14.1, 21.1, 26.5, 28.6, 30.2, 32.4, 33.7; eight methylene carbons as triplets at δ 22.6, 23.1, 24.3, 32.6, 33.9, 34.6, 36.3, 40.5; five CH carbons as doublets at δ 41.0, 43.7, 80.9, 116.4 and 117. 9 and eight singlets six of which saturated sp3 carbons appearing at δ 28.1, 30.1, 33.2, 35.3, 47.2, 51.3 and two as olefin carbon at δ 141.1 and 144.3 ppm and a single carbonyl carbon at δ 179.5 ppm.
3.3. Sodium Dichromate Oxidation of Compound (2)
- A solution of Na2Cr2O7 (5 g) in glacial acetic acid (150 mL) was added slowly during a period of 1 hr to a vigorously stirred solution of product (2) in refluxing CH2Cl2 (50 mL). The mixture was refluxed for 24 hrs and then cooled. Excess dichromate was decomposed with EtOH (50 mL). The solution was concentrated to 1/3 rd volume and the content poured into ice cold water, extracted with ether, washed with water and dried (Na2SO4). Removal of solvent and chromatography of the residue furnished a solid which on crystallization gave the compound (A-2). m.p: 316 ℃, [α] D +16o. The molecular formula of the compound is C30H44O4. IR: 1750 & 1720 cm-1 (γ-lactone ring & a six membered ketone). TNM test is negative. UV: λmax at 250 nm and 290 nm. MS: m/z 468 [M+], the other fragments appeared at m/z 453, 440, 425, 407, 371, 315, 255, 233, 217, 206, 153 and 123 (base peak). PMR: δ 0.99, 1.0 (2 x-CH3), 1.06, 1.19, 1.20, 1.27 (s, 7xt-Me), δ 4.17 (t,1H, J = 3 Hz) due to C-15 lactyl proton, δ 2.39 (dd,J = 7 Hz and 15 Hz) and δ 2.16 (dd, J =7 Hz and 15Hz) for CH2 proton adjacent to the carbonyl group, these two protons couple with a lonely proton at δ 1.89 (t, 1H, J = 7 Hz). 13C NMR: Seven tertiary groups as a quartet at δ 13.3, 16.7, 23.5, 25.8, 30.0, 31.9, 33.3; nine triplets at δ 17.1, 32.2, 29.3, 33.9, 34.5, 35.9, 36.3, 37.2, 40.1; three doublet at δ 34.9, 43.9 and 80.5 and ten singlets six of them being due to sp3 tertiary carbons at δ 28.1, 30.2, 31.5, 36.9, 47.2 and 51.1, two at δ 69.6 and 73.1 are due to carbons attached to the oxygen atom and the two resonating down field carbons at δ 178.8 and 206.1 ppm are due to the carbons of lactone carbonyl and the (C-6) ketone functions.
3.4. Reduction the Compound (2) with Lithium in Ethylenediamine
- Lactone (2) (200 mg) was refluxed in ethylene diamine (150 mL) with lithium metal (200 mg) in presence of nitrogen atmosphere for 3 hrs. After usual work-up and purification by chromatography and repeated crystallization from CHCl3-MeOH, a solid (A-3) was obtained. The molecular formula was analysed C30H50O2; m.p: 265-266 ℃, IR: broad band ranging between 3200-3400 cm-1 for hydroxyl group. MS: m/z 442 [M+], 424 [M+-H2O], 409 [424-CH3], 393 [424- CH2OH] base peak, other fragments appeared at m/z 379, 255, 231, 189, 175, 149, 123, 107, 95, 81, 69. PMR: δ 4.08 (for C-15 proton), δ 3.41 (dd, J = 12 Hz for gem-protons of C-27). Seven tertiary methyl groups appeared at δ 0.97 (2xCH3), 1.0 (3xCH3), 1.02 (CH3), 1.25 (CH3). 13C NMR: Seven quartets for seven CH3 group at 20.0, 21.2, 28.0, 29.0, 30.3, 33.5, and 35.7; twelve triplets at δ 19.9, 20.1, 24.5, 26.3, 28.7, 31.9, 32.5, 35.9, 39.1, 39.7, 48.1 and 64.5 (CH2OH). Three doublets at δ 41.3 43.1, 72.2 (at C-15 as CHOH); eight singlet’s at δ 28.3, 29.1, 33.8, 38.0, 42.2, 43.3, 132.6 and 138.0 ppm (last two signals being due to olefinic carbons).
4. Conclusions
- From the above chemical reactions involving oxidative and reductive transformations of the rearranged product (2), it is evident that usual migration of carbocation to give the most stable olefinic products like β-amyrene[7] or δ-amyrene is restricted to the C-5(10) position due to the presence of lactone ring at C-27(15α) position. It is further concluded that the presence of olefinic double bond at AB- ring juncture (5/10) position facilitates the formation of diol rather than the carboxylic group as observed in the case of parent compound odollactone[7].
ACKNOWLEDGEMENTS
- The authors are thankful to Prof. P Weyerstahl, Institut fur Organische Chemie, Technische Universitat, Berlin, West Germany for the MS, PMR and 13C NMR spectra.