-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2012; 2(3): 63-68
doi: 10.5923/j.ajoc.20120203.05
Utility of Cyclododecanone as Synthon to Synthesize Fused Heterocycles
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLHanafi H. Zoorob , M. S. Elsherbini , Wafaa S. Hamama
Chemistry Department, Faculty of Science Mansoura University, Mansoura 35516, Egypt
Correspondence to: Wafaa S. Hamama , Chemistry Department, Faculty of Science Mansoura University, Mansoura 35516, Egypt.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Ethyl 2-oxocyclododecanecarboxylate (1), and 2-(hydroxyl-methylene)cyclododecanone (7) were used as key intermediates for synthesis of macrocyclic systems incorporating either fused or exocyclic nitrogen heterocycles of different ring sizes. Ring enlargement was also implemented.
Keywords: Cyclododecanone; β-ketoesters; pyrazolones; enaminoketones; fused heterocycles.
1. Introduction
- β-Ketoesters are readily available synthetic building blocks of high importance in organic synthesis. They are used for construction of diverse heterocyclic ring systems belonging to different classes of simple and fused heterocycles[1,2]. Their copper complexes are valuable catalysts in many organic transformations[3,4]. A few of bicyclic or binary systems, including seven-, six- and five-membered heterocycles fused with a dodecamethylene ring, have been synthesized based on cyclododecanone[5,6]. Among these, compounds possessing biological activity and natural compounds, e.g., muscopyridines, have been found[7]. Zoorob et al[8], has been exploring the literature survey of cyclododecanone compounds including their reactivity features. In the present work, we aimed to synthesize a number of heterocycles fused or binary with a dodecamethylene ring starting from cyclododecanone derivatives.
2. Results and Discussion
- Pyrazolones have gained importance as drug substances in pharmaceutical industry in view of their biological importance. For instance, pyrazolones possess antimicrobial, antifungal[9], antimycobacterial[10], antibacterial[11], anti- inflammatory[12], antitumor[13], gastric secretion stimulatory[14], antidepressant[15] and anti-filarial activities[16]. They also serve as precursors for dyes, pigments, pesticides and chelating agents[17]. They also have many industrial applications. Thus, pyrazolone 2 was synthesized by condensation of ethyl 2-oxocyclodode- canecarboxylate (1) with phenylhydrazine. The structure of 2 was supported by IR, 1H NMR and mass spectra. Its IR spectrum displays a strong absorption at υ = 3277 (NH) and at 1696 cm-1 (γ-lactam carbonyl group). Its 1H NMR spectrum revealed that it may exist as a mixture of two tautomers 2a and 2b, where it displays a singlet signal for 1H at δ 6.4 ppm indicating NH group (structure 2b) and a multiplet signal for 1H at δ 2.65 ppm indicating the proton at C4 (structure 2a) besides the rest of bands corresponding to 5 aromatic protons at δ 7.1-7.8 ppm and 16 aliphatic protons at 1.1-1.5[18].Also, the isoxazolone 3 was simply synthesized by reaction of the ketoester 1 and hydroxylamine hydrochloride in refluxing methanol containing pyridine. The structure of 3 was attested by IR, 1H NMR and MS spectra. Its 1H NMR spectrum displays the main important signals that correspond to the proton adjacent to the carbonyl group as a multiplet at δ 2.55 (1H), besides the other twenty protons as quartet at δ 2.35 (2H) and multiplet at δ 1.1-1.7 ppm (18H). Moreover, the mass spectrum of 3 was in a good agreement with its structure where it displays a molecular ion peak with m/z 223 (24.3%).Furthermore, aminopyrimidines are important biologically active agents such as antiplatelet[19] and as VEGF-R2 inhibitors[20] and they are also important synthetic intermediates for fused heterocycles[21,22]. This enthused us to synthesize 2-amino-5,6,7,8,9,10,11,12,13,14-decahydrocyclododeca[d]pyrimidin-4(3H)-one (4) via treatment a mixture of the ketoester 1 and guanidine nitrate with sodium ethoxide in ethanol. The 1H NMR of 4 spectrum displays a signal corresponding to eight methylene groups as multiplet at δ 1.1-1.6 (16H), a triplet signal at δ 2.25 (4H) corresponding to two methylenes neighboring to the double bond, a singlet signal at δ 6.5 ppm (2H) corresponding to (NH2) group and a singlet signal at δ 8.08 ppm (1H) corresponding to (NH) group.Furthermore, 2H-chromene-2-ones (coumarines) continue to be investigated because of their importance to medicinal chemists due to a variety of biological activities. Associated with the coumarine scaffold are antibacterial, antiviral, anticancer activity as well as inhibition of platelet aggregation and inhibition of steroid 5α-reductase. Coumarines are used in drug and pesticidal preparations; they have also found applications as photosensitizers, fluorescent and laser dyes[23,24]. Because of the significant activities of these molecules we have prepared the coumarin 5 through condensation of ethyl 2-oxocyclo-dodecane carboxylate (1) with m-cresol under Von Pechmann reaction conditions[25]. The chemical structure of 5 was proved by IR and mass spectra. Its IR spectrum displays a peak at υ 1706 cm-1 due to lactone carbonyl group. In addition, its mass spectrum was perfectly consistent with its chemical structure where it displays a molecular ion peaks at m/z 298 (20%).Treatment of 1 with hydrazoic acid generated in situ from sodium azide and concentrated sulfuric acid in chloroform at low temperature furnished the azacyclotridecane derivative 6 in high yield. The IR spectrum of compound 6 showed a strong absorption at υ = 3316 cm-1 due to (NH) group. Also, the main characteristic features of the 1H NMR spectrum of 6 was the presence of a signal at δ 6.2 (1H, singlet) which is assigned to the proton of (NH) group, the signal at δ 4.7 (1H) which is assigned to the proton on the carbon located between (NH) and ester groups and the multiplet signal at δ 4.2 ppm (2H) which is assigned to the methylene group next to the carbonyl group. The assignment of the NH group between the CO groups and the substituted carbon atoms in compound 6 is based on the previously reported studies by Hamama et al[26] and Schmidt et al[27].
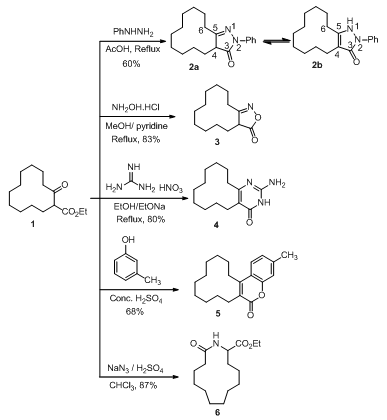 | Scheme 1. Synthesis of different heterocycles fused or including dodecamethylene ring |
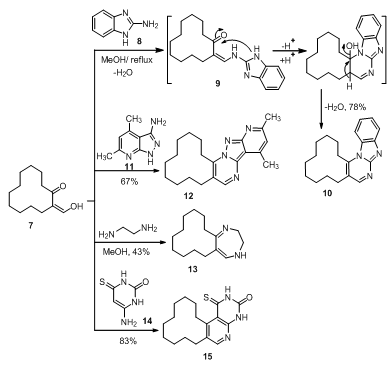 | Scheme 2. Reaction of 2-(hydroxymethylene)cyclododecanone (7) with different primary amines |
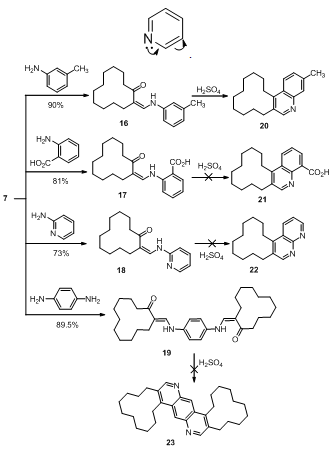 | Scheme 3. Synthesis of enaminoketones 16-19 and trials to synthesize their fused heterocycles 20-23 under concentrated sulfuric acid influence |
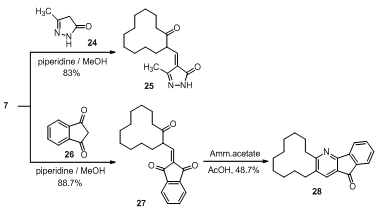 | Scheme 4. Condensation of 2-(hydroxymethylene)cyclododecanone (7) with active methylene compounds |
3. Conclusions
- In this paper, compounds ethyl 2-oxocyclododecane- carboxylate (1) and 2-(hydroxymethylene)cyclododecanone (7) were used as starting compounds for the synthesis of the target compounds. Thus, compound 1 reacted with different nucleophiles at different reactions conditions to give pyrazolone 2, isoxazolone 3 pyrimidinone 4, coumarin 5 and azacyclotridecane 6 derivatives. Furthermore, compound 7 followed condensation reactions with bifunctional nucleophiles such as benzo[d]imidazole 8, pyrazolo[3,4-b]pyridine 11, ethylene diamine and 6-aminothiouracil 14 to give pyrimidine 10, pyrazolopyrimidine 12, diazepine 13 and pyridopyrimidine 15, respectively. Moreover, enaminones 16-19 were obtained by interaction of 7 with the appropriate amines. Finally, the base catalyzed condensation of 7 with active methylene compounds afforded the corresponding alkylidenes 25 and 27, respectively.
4. Experimental
- All melting points are in degree centigrade (uncorrected) and were determined on Gallenkamp electric melting point apparatus. The IR spectra were recorded (KBr) on a Mattson 5000 FTIR spectrophotometer at Microanalytical Unit, Faculty of Science; Mansoura University. The 1H NMR data were obtained in CDCl3 or DMSO using TMS as internal standard and chemical shifts were reported in ppm (δ) downfield from internal TMS. The 1H NMR spectra of compounds 2-4, 6 and 10 were measured on a JEOL (500 MHz) spectrometer; Alexendria University. The mass spectra of compounds 5, 6, 12, 15, 20 and 28 were carried out on GCMS-QP 1000 EX Shimadzu Japan (Gas Chromatography-Mass spectrometer), National Research Center, Dokki, Giza, Egypt. The 13C NMR spectrum for compound 17 (125 MHz) was performed on a Bruker Avance 500 spectrometer at Bioorganic Chemistry department, Saarland Saarbrüken University, Germany. The Mass spectra of compounds 2, 10, 13, 16-19, 25 and 27 were recorded on a Finnigan MAT 212 mass spectrometer instrument, Micro analytical unit Cairo University. Compounds 3 and 4 were recorded on 70 eV with a Varian MAT 311 instrument. Reactions were monitored by thin layer chromatography (TLC) using EM science silica gel coated plates with visualization by irradiation with ultraviolet lamp. The ethyl2-oxocyclododecanecarboxylate (1), and2-(hydroxymethylene)cyclododecanone (7) were prepared as previously cited in literature according to reported methods[30,31].N-Phenyl-1,2,4,5,6,7,8,9,10,11,12,13-dodecahydro-cyclododecapyrazol-3-one (2). A mixture of 1 (1 g, 3.94 mmol) and phenyhydrazine (0.425 g, 0.387 mL, 3.94 mmol) was refluxed in acetic acid (20 mL, 50%) for 3 h, poured into ice-cold water while stirring, whereby a yellow precipitate was formed, filtered off and air dried, recrystallized from ethanol to furnish 2. Yield 60%; mp 178-180°C; IR (KBr) ύmax. cm-1: 3277, 2931, 2858, 1697; 1H NMR (500 MHz, DMSO) δ ppm: 1.1-1.5 (16 H, m), 1.75 (2H, t, J= 7.65 MHz), 2.05 (1H, m), 2.35 (1H, m), 2.65 (1H, m), 6.4 (1H, s), 7.15 (1H, t, J= 7.65 MHz), 7.4 (2H, t, J = 7.65 MHz), 7.75 (2H, d, J= 7.65 MHz); MS (EI, 70 ev): m/z (%): 298 (M+, 7.24), 297 ([M-H] +, 15), 77 (100); Anal. Calcd. for C19H26N2O (298.24): C, 76.47; H, 8.78; N, 9.39; Found: C, 76.66; H, 8.69; N, 9.32.4,5,6,7,8,9,10,11,12,13-Decahydrocyclododeca[c]isoxazol-3(3aH)-one (3). A mixture of 1 (0.635 g, 2.5 mmol), hydroxylamine hydrochloride (0.174 g, 2.5 mmol) and pyridine (2 mL) was refluxed in methanol (10 mL) for 5 h, left to cool, poured into ice-cold water and acidified with conc. HCl whereby an oily product was formed. The oil was separated and triturated with petroleum ether to give a white precipitate. Recrystallization from a mixture of ethanol and DMF (10:1) afforded 3. Yield 83%; mp 106-110°C; IR (KBr) ύmax. cm-1: 2930, 2853, 1673; 1H NMR (500 MHz, CDCl3) δ ppm: 1.1-1.7 (18H, m), 2.35 (2H, q, J= 6.90 MHz), 2.55 (1H, m); MS (EI, 70 ev): m/z (%): 223 (M+, 24.3), 55 (100); Anal. Calcd. for C13H21NO2 (223.19): C, 69.92; H, 9.48; N, 6.27; Found: C, 69.74; H, 9.75; N, 6.21.2-Amino-5,6,7,8,9,10,11,12,13,14-decahydrocyclododeca[d]pyrimidin-4(3H)-one (4). Guanidine nitrate (0.61 g, 5 mmol) was added to an ethanolic solution of sodium ethoxide[prepared by dissolving sodium (0.23 g, 10 mmol) in ethanol (30 mL)] then ketoester 1 (1.27 g, 5 mmol) was added. The reaction mixture was heated under reflux over steam bath for 8 h, left to cool whereby a white precipitate was formed and filtered off. Recrystallization from a mixture of ethanol and DMF (10:1) furnished compound 4. Yield 80%; mp 260-262°C; IR (KBr) ύmax. cm-1: 2934, 2857, 3408, 3308, 3134, 2928, 2855; 1H NMR (500 MHz, DMSO) δ ppm: 1.0-1.6 (16H, m), 2.25 (4H, t, J= 6.10 MHz), 6.5 (2H, s), 8.08 (1H, s); MS (EI, 70 ev): m/z (%): 249 (M+, 20.9), 139 (100); Anal. Calcd. for C14H23N3O (249.22): C, 67.43; H, 9.30; N, 16.85 Found: C, 67.61; H, 9.22; N, 16.79.3-Methyl-7,8,9,10,11,12,13,14,15,16-decahydro[1]cyclododeca[c]chromen-6-one (5). A mixture of the ketoester 1 (1.28 g, 5 mmol) and m-cresol (0.54 g, 5 mmol) was heated in sulfuric acid (10 mL, 75%) over steam bath for 1.5 h. Then, left to cool and poured into crushed ice whereby a white precipitate was formed, filtered off and dried in a dissicator (over anhydrous calcium chloride) then recrystallized from ethanol-water mixture (15: 2) to give 5. Yield 68%; mp 75-77°C; IR (KBr) ύmax. cm-1: 2933, 2858, 1706; MS (EI, 70 ev): m/z (%): 298 (M+, 20), 55 (100); Anal. Calcd. for C20H26O2 (298.23): C, 80.50; H, 8.78; Found: C, 80.37; H, 8.82.13-Oxo-azacyclotridecane-2-carboxylic acid ethyl ester (6). Ketoester 1 (1.28 g, 5 mmol) was added with stirring to a pre-cooled (salt-ice bath) mixture of (30 mL) chloroform and sulfuric acid (10 mL, 80%) then sodium azide (0.325 g, 5 mmol) was added portion wise over 1 h with stirring in salt-ice bath. The mixture was then heated over steam bath for 10 min; stirring was continued for additional 4 h at room temperature. The reaction mixture was then poured into ice-cold water and basified with concentrated ammonia solution whereby a gelatinous white precipitate was formed which then extracted with chloroform (3x20 mL). The combined chloroform extract was dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure to afford a white precipitate recrystallized from a mixture of methanol and DMF (10:1) to produce compound 6. Yield 87%; mp 140-142°C; IR (KBr) ύmax. cm-1: 3316.9, 2921, 2858, 1741, 1708; 1H NMR (500 MHz, CDCl3) δ ppm: 1.29-1.30 (17H, m), 1.57 (2H, m), 2.07 (2H, m), 2.20 (2H, m), 4.2 (2H, m), 4.7 (1H, m), 7.25 (1H, s); MS (EI, 70 ev): m/z (%): 269 (M+, 24), 196 (100); Anal. Calcd. for C15H27NO3 (269.24): C, 66.88; H, 10.10; N, 5.20; Found: C, 66.77; H, 10.02; N, 5.17.Benzimidazo[1,2-a]-5,6,7,8,9,10,11,12,13,14-decahydro [1]cyclododeca[d]pyrimidine (10). Compound 7 (0.53 g, 2.25 mmol) and 2-aminobenzimidazol (8) (0.3 g, 2.25 mmol) was completely dissolved in methanol (20 mL). The reaction mixture was refluxed for about 3 h after that it was left to cool overnight. Pale yellow crystals were separated, filtered off and air dried. Recrystallization from ethanol gave 10. Yield 78%; mp 230°C; IR (KBr) ύmax. cm-1: 2921, 2860, 1633; 1H NMR (500 MHz, CDCl3) δ ppm: 1.3-1.6 (12H, m), 1.8 (2H, m), 2.0 (2H, m), 2.8 (2H, t, J= 7.60 MHz), 2.9 (2H, t, J= 7.65 MHz), 7.36 (1H, t, J= 7.60 MHz), 7.52 (1H, t, J= 7.65 MHz), 7.93 (1H, d, J= 8.40 MHz), 8.0 (1H, d, J= 8.40 MHz), 8.68 (1H, s); MS (EI, 70 ev): m/z (%): 307 (M+, 100); Anal. Calcd. for C20H25N3 (307.24): C, 78.14; H, 8.24; N, 13.67; Found: C, 77.93; H, 8.12; N, 13.58.Synthesis of 13,15-dimethyl-1,2,3,4,5,6,7,8,9,10- deca- hydrocyclododeca[e]pyrido[2',3':3,4]pyrazolo[1,5-a] pyrimidine (12). A mixture of ketoaldehyde 7 (0.45 g, 2.1 mmol) and the pyrazolopyridine derivative 11 (0.35 g, 2.1 mmol) was dissolved in a mixture of MeOH (20 mL) and DMF (5 mL), The reaction mixture was then heated under reflux for 6 h, left to cool overnight, poured into crushed ice to afford a yellow precipitate which was collected by filtration under vacuum and air dried. Recrystallization from a mixture of ethanol and DMF (10:1) afforded 12. Yield 67%; mp 130-133°C; 337 ([M+H] +, 10.15), 336 (M+, 26), 55 (100); Anal. Calcd. for C21H28N4 (336.27): C, 74.96; H, 8.39; N, 16.65; Found: C, 74.85; H, 8.32; N, 16.61.(1Z,5Z)-3,4,6,7,8,9,10,11,12,13,14,15-Dodecahydro-2H-[1]cyclododeca[e][1,4]diazepine (13). A mixture of ketoaldehyde 7 (0.53 g, 2.5 mmol) and ethylene-diamine (0.15 g, 0.17 mL, 2.5 mmol) was refluxed in methanol (20 mL) for 2.5 h, left to cool overnight, filtered off and air dried. Recrystallization from ethanol afforded diazepine 13. Yield 43%; mp 260-264℃; IR (KBr) ύmax. cm-1: 3300, 2931, 2858, 1643; MS (EI, 70 ev): m/z (%):234 (M+, 3.25), 235[(M+H) +, 14.9], 222 (100); Anal. Calcd. for C15H26N2 (234.24): C, 76.87; H, 11.18; N, 11.95; Found: C, 76.78; H, 11.15 N, 11.89.Synthesis of 1-thioxo-1,2,7,8,9,10,11,12,13,14,15,16- dodecahydrocyclododeca[4,5]pyrido[2,3-d]pyrimidin-3 (4H)-one (15). A mixture of ketoaldehyde 7 (0.53 g, 2.5 mmol) and 6-aminothiouracil (14) (0.36 g, 2.5 mmol) was refluxed in DMF (15 mL) for 4.5 h, then poured into crushed ice whereby a yellow precipitate was formed, filtered off and air dried then recrystallized from a mixture of ethanol and DMF (10:1) to afford 15. Yield 83%; mp 148-150℃; IR (KBr) ύmax. cm-1: 3426, 3322, 2925, 2858, 1625; MS (EI, 70 ev): m/z (%): 317 (M+, 32), 98 (100); Anal. Calcd. for C17H23N3OS (317.28): C, 64.32; H, 7.30; N, 13.24; Found: C, 64.21; H, 7.24; N, 13.18.2[N-(m-Toluidino)methylene]cyclododecanone (16). Ketoaldehyde 7 (1.05 g, 5 mmol) was added to m-toluidine (0.54 g, 5 mmol) in methanol (20 mL). The reaction mixture was refluxed for 2 h, left to cool whereby a yellow precipitate was formed, filtered off and air dried then recrystallized from ethanol to give 16. Yield 90%; mp 185-187℃; IR (KBr) ύmax. cm-1: 3253, 2926, 2853, 1636; MS (EI, 70 ev): m/z (%): 297 (M+-2, 57), 298 (M+-1, 12), 187 (100); Anal. Calcd. for C20H29NO (299.26): C, 80.22; H, 9.76; N, 4.68; Found: C, 80.01; H, 9.59; N, 4.57.2-[(2-Oxocyclododecylidene)methylamino]benzoic acid (17). Ketoaldehyde 7 (1.05 g, 5.0 mmol) was refluxed with a methanolic solution (25 mL) of anthranilic acid (0.68 g, 5.0 mmol) for 1 h. The reaction mixture was left to cool, filtered off and air dried. Recrystallization from ethanol furnished 17. Yield 81%; mp 215-217℃; 1H NMR (500 MHz, DMSO) δ ppm: 1.0-1.65 (16H, m), 2.35 (2H, m), 2.7 (2H, m), 4.04 (1H, s), 6.95 (1H, d, J= 7.65 MHz), 7.25 (1H, s), 7.65 (1H, t, J= 7.65 MHz), 7.9 (1H, t, J= 7.65 MHz), 8.05 (1H, d, J= 7.65 MHz), 10.7 (1H, s); 13C NMR (125 MHz, DMSO): δ ppm: 20.97, 22.42, 22.51, 22.87, 23.70, 23.97, 24.80, 25.18, 25.52, 28.71, 117.01, 125.03, 127.69, 128.15, 134.16, 135.70, 138.35, 155.08, 178.42, 179.96; 13C NMR (DEPT 135, T= 60℃, DMSO): indicates a presence of ten CH2 and one CH groups; MS (70 eV): m/z = 329 (M+, 49), 137 (100); Anal. Calcd. for C20H27NO3 (329.24): C, 72.92; H, 8.26; N, 4.25; Found: C, 72.85; H, 8.22; N, 4.21.2-[(Pyridin-2-ylamino)methylene]cyclododecanone (18). Ketoaldehyde 7 (1.05 g, 5 mmol) was added to 2-aminopyridin (0.47 g, 5.0 mmol) in methanol (20 mL) and allowed to react for about 3 h. The reaction mixture was left to cool over night whereby a crystalline white precipitate was formed, filtered off and air dried. Recrystallization from ethanol produced 18. Yield 73%; mp 180-182℃; IR (KBr) ύmax. cm-1: 3319, 2926, 2856, 1654; 1H NMR (500 MHz, DMSO) δ ppm: 1.0-1.65 (16H, m), 2.35 (2H, m), 2.7 (2H, m), 4.04 (1H, s), 6.95 (1H, t, J= 6.10 MHz), 7.25 (1H, s), 7.65 (1H, d, J= 7.65 MHz), 7.9 (1H, t, J= 6.10 MHz), 8.05 (1H, d, J= 8.03 MHz); MS (EI, 70 ev): m/z (%): 286 (M+, 45), 145 (100); Anal. Calcd. for C18H26N2O (286.24): C, 75.48; H, 9.15; N, 9.78; Found: C, 75.39; H, 9.07; N, 9.71.Bis-2-[N,N’-(p-phenylenediaminomethylene)]cyclododecanone (19). Ketoaldehyde 7 (0.53 g, 2.5 mmol) was mixed with p-phenylenediamine (0.14 g, 1.25 mmol) in methanol (20 mL). The reaction mixture was refluxed for 1 h then poured into ice cold water. A greenish precipitate was formed, filtered under suction and air dried. Recrystallization from ethanol gave 19. Yield 89.5%; mp 110℃; IR (KBr) ύmax. cm-1: 3340, 2929, 2856, 1635; MS (EI, 70 ev): m/z (%): 492 (M+, 100); Anal. Calcd. for C32H48N2O2 (492.43): C, 78.00; H, 9.82; N, 5.69; Found: C, 78.11; H, 9.77; N, 5.64.3-Methyl-7,8,9,10,11,12,13,14,15,16-decahydro-[1]cyclododeca[c]quinoline (20). A mixture of enaminoketone 16 (0.3 g, 1 mmol) and concentrated sulfuric acid (5 mL) was heated over a steam bath for 3 h. Poured into crushed ice then basified with ammonium hydroxide whereby a white precipitate was formed, filtered off and recrystallized from methanol to give 20. Yield 53%; mp 108-110℃; IR (KBr) ύmax. cm-1: 2923, 2877, 1633; MS (EI, 70 ev): m/z (%): 281 (M+, 18), 98 (100); Anal. Calcd. for C20H27N (281.24): C, 85.35; H, 9.67; N, 4.98; Found: C, 85.27; H, 9.63; N, 4. 92.3-Methyl-4-((2-oxocyclododecyl)methylene)-1H-pyrazol-5(4H)-one (25). To a mixture of ketoaldehyde 7 (1.05 g, 5.0 mmol) and pyrazolone 24 (0.73 g, 5 mmol) in methanol (20 mL) a few drops of piperidine were added. The reaction mixture was heated under reflux for 2 h. Removal of solvent under vacuum affording a reddish precipitate which upon recrystallization from methanol afforded 25. Red crystals; yield 83%; mp 231-234℃; IR (KBr) ύmax. cm-1: 3212, 2920, 2859, 1706, 1592; MS (EI, 70 ev): m/z (%): 290 (M+, 5.77), 206 (100); Anal. Calcd. for C17H26N2O2 (290.24): C, 70.31; H, 9.02; N, 9.65; Found: C, 70.22; H, 9.00; N, 9.61.2-((2-Oxocyclododecyl)methylene)-2H-indene-1,3-dione (27). To a mixture of ketoaldehyde 7 (1.05 g, 5 mmol) and 1,3-indandione (26) (0.73 g, 5 mmol) in methanol (20 mL), a few drops of piperidine were added. The reaction mixture was heated under reflux for 3 h, poured into crushed ice and acidified with HCl. A brown precipitate was formed, filtered off, air dried and recrystallized from ethanol to give 27. Yield 88.7%; mp 120-124℃; IR (KBr) ύmax. cm-1: 2929, 2858, 1708, 1629; MS (EI, 70 ev): m/z (%): 338 (M+, 7.26), 55 (100); Anal. Calcd. for C22H26O3 (338.23): C, 78.07; H, 7.74; Found: C, 78.00; H, 7.70.Indenopyridine derivative (28). Alkylidine 27 (0.6 g, 1.8 mmol) was dissolved in glacial acetic acid (15 mL) containing (2 g) ammonium acetate, the reaction mixture was then heated under reflux for 5 h. Left to cool, then poured into crushed ice whereby, a grey precipitate was formed, filtered off, dried and recrystalized from a mixture of ethanol and DMF (10:1) to afford 28. Yield 48.7%; mp 134-136℃; IR (KBr) ύmax. cm-1: 2925, 2854, 1708; MS (EI, 70 ev): m/z (%): 319 (M+, 35), 320[(M+H)+, 28], 209 (100); Anal. Calcd. for C22H25NO (319.23): C, 82.72; H, 7.89; N, 4.38; Found: C, 82.66; H, 7.83; N, 4.32.