Sridhar Poloju 1, Kishore Cholkar 2, Gilles K. Kouassi 1, J. David Van Horn 3, K.S. Rangappa 4, Netkal M. Made Gowda 1
1Department of Chemistry, Western Illinois University, One University Circle, Macomb, IL 61455, USA
2Division of Pharmaceutical Sciences, School of Pharmacy, University of Missouri-Kansas City, 2464 Charlotte Street, Kansas City, MO 64108, USA
3Department of Chemistry, University of Missouri-Kansas City, 5110 Rockhill Road, Kansas City, MO 64110, USA
4Department of Studies in Chemistry, Manasagangothri, University of Mysore, Mysore 570 006, INDIA
Correspondence to: Netkal M. Made Gowda , Department of Chemistry, Western Illinois University, One University Circle, Macomb, IL 61455, USA.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Abstract
The kinetics of oxidation of lactic acid (LA) by manganese(III) in sulfuric acid solutions at 293 K has been studied. A solution of the mild oxidant, Mn(III) sulfate, in aqueous sulfuric acid medium was prepared by a standard electrochemical method. The oxidation reaction was monitored by spectrophotometry at a fixed wavelength (λmax = 491 nm), varied temperature, and varied solution conditions. The Mn(III)-LA reaction stoichiometry of 4:1 was determined and the products were characterized. The experimental rate law is: rate = kobs [Mn(III)][LA]x[H+]y, where x, and y are fractional orders. The effects of varying ionic strength, solvent composition (dielectric constant), acid, and the reduction product, Mn(II), on the rate of the reaction were investigated. Activation parameters evaluated using Arrhenius and Eyring plots suggest the occurrence of an entropy controlled reaction. A mechanism consistent with the observed kinetic data has been proposed. A rate law has been derived based on the mechanism.
Keywords:
Kinetics, Oxidation, Lactic Acid, Manganese(III), Mechanism, Rate Law
1. Introduction
Lactic acid is an important biochemical and industrial molecule; it is a useful feedstock used in the generation of aldehydes and ketones via oxidative methods. Colored manganese ions are used industrially as pigments and in its higher oxidation states, Mn is used as an oxidant.[1] For chemical transformations, electrochemically or chemically generated Mn(III) is used as a mild oxidizing agent and has special importance due to its biological relevance.[1] Anaerobic oxidation of lactic acid and its products has been reported. An aerobic oxidation mechanism of lactic acid to pyruvic acid has also been reported.[2] Oxidations of lactic acid by oxidants such as chromic acid,[3] chloramine-B, pyridinium chlorochromatein in acidic media,[4] water soluble manganese dioxide,[5] and permanganate ion in acidic solutions[6] have been reported. Manganese(II) ions function as cofactors for a number of enzymes whereas Mn is a required trace mineral for essentially all living organisms. There is a pronounced importance of manganese in growth, bone development, reproduction, and in the central nervous system. Manganese accumulates in mitochondria and is essential for their function. Manganese is also important in photosynthetic oxygen evolution in plants.[1] There are several reports of Mn(III) oxidation of various substrates including α-hydroxy acids,[7] peptide polymers,[8] and organic substrates such as pyruvic acid[9] in perchlorate,[7] acetate,[7] sulfate,[9] or pyrophosphate media.[1] Some of these oxidation mechanisms involve free radicals formed in situ while the LA oxidation by Mn(III) does not involve free radicals in the mechanism. The present study of the oxidation of lactic acid by Mn(III) in sulfuric acid solutions has been initiated to characterize the one-electron oxidation behavior of Mn(III) and its action as a carbon-carbon bond cleaving agent.
2. Experimental
Lactic acid (ACROS Co., USA) (purity 99.5%) was used as supplied to prepare stock aqueous solutions. Aqueous solutions of Mn(II) sulfate (Aldrich Chemical Co.) were prepared. All solutions were prepared using analytical grade chemicals and doubly distilled water. The reaction was followed under pseudo-first-order conditions by monitoring the absorbance of Mn(III) with time at λmax = 491 nm. The course of the reaction was monitored using Shimadzu UV-visible model 1601 spectrophotometer fitted with a temperature controlling unit or thermostat. 1H-NMR spectra were obtained on a JEOL 300 MHz or Varian INOVA 400 MHz spectrometer. IR spectra were collected on a Shimadzu FTIR 8400S spectrophotometer.
2.1. Electrochemical Preparation of Manganese(III)
A solution of Mn(III) sulfate was prepared using a standard anodic oxidation of 0.200 M solution of Mn(II) sulfate in 3.00 M H2SO4[1,]. The electrolysis was performed in an undivided cell with a platinum foil anode (generation area 4.0 cm2) and a thin platinum spiral cathode (effective area 0.2 cm2). The experiment was carried out at a cell voltage of approximately 3 V and a current density of 2 mA cm-2. The solution was continuously stirred and the electrolysis continued until a Mn(III) concentration of approximately 0.030 M was generated. The exact concentration of the pink manganese(III) sulfate solution was determined iodometrically. Further electrolysis resulted in the precipitation of Mn(III) sulfate due to its solubility limit. The manganese(III) sulfate solution was maintained with a known excess amount of Mn(II) sulfate to suppress the following disproportionation reaction: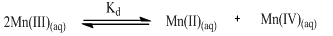 | (1) |
The prepared solution appeared to be stable for more than a month at [H+] ≥ 3.0 M and ambient laboratory temperature. However, the prepared Mn(III) solution was iodometrically standardized before use in experiments. The UV-visible spectrum of the manganese(III) sulfate solution shows its λmax at 491 nm. The standard redox potential E′0 of Mn(III)/Mn(II), reflecting the oxidizing power of the oxidant, generally decreases upon complexation. The E′0 values measured at specified experimental conditions were as reported previously[1,10].
2.2. Kinetic Measurements
Kinetic runs were performed under pseudo-first-order conditions with a large excess of the substrate, lactic acid (LA), as compared to the oxidant, Mn(III), at 20℃. For each run, requisite amounts of standard LA solution and H2SO4 (to maintain a constant acid strength) were mixed in a 50 mL volumetric flask. To this solution, a measured amount of pre-equilibrated Mn(III) solution was added to give the known initial concentration. Enough water was added to the flask to make-up the final volume to 50.0 mL. The course of the reaction was monitored spectrophotometrically in a 1 cm cuvette by measuring the absorbance of Mn(III) (λmax = 491 nm) at regular time intervals for five hours. For each run, absorbance-time data were obtained. Linear pseudo-first- order plots of ln (Abs) vs. time were obtained, where the slope gave the first-order rate constant, k′ or kobs. The calculated k′ values were reproducible within ± 3% error. Each k′ value is an average of two runs.
2.3. Reaction Stoichiometry
Reaction mixtures containing varying mole ratio of LA-to- Mn(III) in the presence of 0.35 M sulfuric acid were kept with occasional stirring in a waterbath at 50℃ for varying time intervals up to 48h. A higher temperature of 50℃ instead of the usual 20℃ was used to permit a faster reaction. Ten mL aliquots of the reaction mixture were withdrawn periodically and the concentration of the unreacted Mn(III) was determined iodometrically at each time interval. A suitable control containing the Mn(III) solution without LA was also run.For example, a blank was prepared as follows: in an Erlenmeyer flask, 75 mL of 3.38 10- Mn(III) solution in 0.350 M H2SO4 was kept with occasional stirring in a waterbath at 50℃. At time intervals of 3, 6, 12, 24, and 48 h, 10-mL aliquots of the blank Mn(III) solution were withdrawn and iodometrically titrated against a standardized 0.0136 N Na2S2O3 solution using 1% starch solution as the indicator near the end point.A typical reaction mixture was prepared as follows: to 75 mL of 3.38 10-2 M Mn(III) solution in 0.350 M H2SO4 taken in a stoppered Erlenmeyer flask in a water at 50℃, 1.44 mL of 0.200 M LA solution was added and the solution mixture was occasionally stirred. At time intervals of 3, 6, 12, 24, and 48 h, 10-mL aliquots of the reaction mixture were titrated determine the unreacted [Mn(III)], as mentioned before.The preliminary results of the reaction that four moles of Mn(III) were consumed per mole of LA. The 4:1 stoichiometric reaction is presented in Scheme I.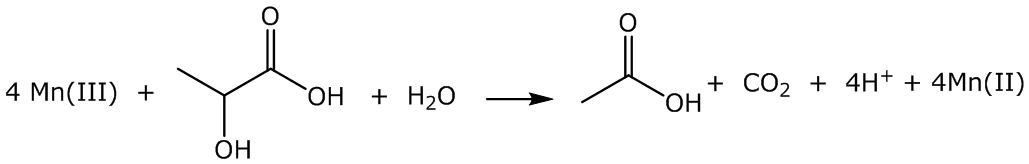 | Scheme I. stoichiometric reaction for the lactic acid oxidation by manganese(III) |
2.4. Product Analysis
Reaction mixtures containing [Mn(III)]o >> [LA]o in 0.35 M H2SO4 were stirred in a water at 50℃ for 48 h. The solvent was removed using a rotary evaporator under reduced pressure. The residue was extracted using diethyl ether-water mixtures (1:1, v/v) in a separatory funnel.[10] The relatively less polar organic layer contained acetic acid and a minor amount of unreacted LA while the aqueous layer contained Mn(II) and Mn(III) sulfate. The organic layer was concentrated with a rotary evaporator. The product, acetic acid, was identified by 1H-NMR (in C6D6) and IR (KBr pellet) spectroscopy by comparing the data with those of an authentic sample. The evolution of CO2 was tested by a conventional lime-water test[11,12]. The presence of Mn(II) in the reaction mixture was indicated by a hypsochromic shift in the λmax of Mn(III)from 491 nm to 474 nm.
2.5. Test for Free Radicals
Acrylonitrile or freshly prepared 10% acrylamide solution, under the inert nitrogen atmosphere, was added to the LA-Mn(III) reaction mixture in 0.35 M H2SO4 to initiate polymerization by free radicals formed in situ [1,10]. The reaction flasks covered with aluminum foils were kept in the dark to prevent photochemical reactions. No turbidity or polymer precipitation was observed indicating the absence of free radicals in the reaction mixture. Suitable controls without LA or Mn(III) were also run simultaneously.
3. Results and Discussion
Under pseudo-first-order conditions with the [LA]o >> [Mn(III)]o at 20℃, the values of the observed rate constant remained the same with varying [Mn(III)]o (Table 1). A linear plot of ln (Abs) vs. time showed the first-order dependence of the rate on [Mn(III)]. At constant [Mn(III)]o, [H+], [LA]o, [Mn(II)]o, and temperature, the reaction rate increased with an increase in [LA]o (Table 1). A plot of ln kobs vs. ln [LA]o was linear with a slope of 0.254 (Fig. 1), indicating a fractional-order dependence of the rate on [LA]o. Varied concentrations of Mn(II), added sodium sulfate or perchlorate had a negligible effect on the rate (Tables 1 and 2). As a result, a constant ionic strength was not maintained in other kinetic runs.| Table 1. Effect of varied [Mn(III)], [Mn(II)], [LA], and [H2SO4] on the rate of oxidation of lactic acid at 293 K |
| | [Mn(III)] (10-4 M) | [LA] (10-2 M) | [Mn(II)] (10-1 M) | [H2SO4] (10-1 M) | kobs (10-4, s-1) | | 2.50 | 2.00 | 2.00 | 3.50 | 6.00 | | 5.00 | 2.00 | 2.00 | 3.50 | 6.75 | | 7.50 | 2.00 | 2.00 | 3.50 | 5.69 | | 10.0 | 2.00 | 2.00 | 3.50 | 6.58 | | 12.5 | 2.00 | 2.00 | 3.50 | 6.16 | | 10.0 | 1.00 | 2.00 | 3.50 | 4.49 | | 10.0 | 2.00 | 2.00 | 3.50 | 5.29 | | 10.0 | 3.00 | 2.00 | 3.50 | 5.71 | | 10.0 | 4.00 | 2.00 | 3.50 | 6.14 | | 10.0 | 5.00 | 2.00 | 3.50 | 6.94 | | 10.0 | 10.0 | 2.00 | 3.50 | 8.39 | | 10.0 | 20.0 | 2.00 | 3.50 | 9.22 | | 10.0 | 2.00 | 2.00 | 3.50 | 5.29 | | 10.0 | 2.00 | 4.00 | 3.50 | 5.59 | | 10.0 | 2.00 | 5.00 | 3.50 | 5.22 | | 10.0 | 2.00 | 6.00 | 3.50 | 5.03 | | 10.0 | 2.00 | 8.00 | 3.50 | 4.67 | | 10.0 | 2.00 | 10.0 | 3.50 | 4.88 | | 10.0 | 2.00 | 2.00 | 1.50 | 3.88 | | 10.0 | 2.00 | 2.00 | 2.00 | 4.43 | | 10.0 | 2.00 | 2.00 | 3.00 | 4.56 | | 10.0 | 2.00 | 2.00 | 4.00 | 4.95 | | 10.0 | 2.00 | 2.00 | 5.00 | 5.16 | | 10.0 | 2.00 | 2.00 | 7.50 | 6.15 | | 10.0 | 2.00 | 2.00 | 10.0 | 6.80 |
|
|
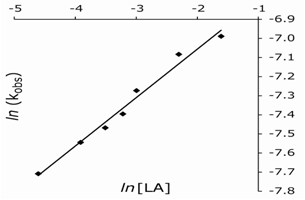 | Figure 1. A plot of ln kobs vs. ln [LA] |
| Table 2. Effect of added sodium salts or MeOH on the rate at 293 K. [Mn(III)]o = 1.00 × 10-3M; [LA]o = 2.00 × 10-2M; [Mn(II)]o = 2.00 × 10-1M; [H2SO4] = 3.50 × 10-1M |
| | Added compound | Concentration | kobs (10-4 s-1) | | | | | %[MeOH] (v/v) | 5.00 | 5.53 | | | 10.0 | 5.85 | | | 15.0 | 6.65 | | | 20.0 | 6.94 | | | 25.0 | 7.56 | | 10 [Na2SO4] (M) | 0.00 | 5.29 | | | 0.50 | 5.43 | | | 1.00 | 5.76 | | | 2.00 | 5.94 | | | 5.00 | 6.11 | | 10 [NaClO4] (M) | 0.00 | 5.29 | | | 0.50 | 4.83 | | | 1.00 | 4.81 | | | 2.00 | 4.80 | | | 5.00 | 4.47 |
|
|
At constant [Mn(III)]o, [LA]o, [Mn(II)]o, and temperature, the reaction rates were measured at varying sulfuric acid concentrations. The rate of reaction increased with increased acid concentration. A plot of ln kobs vs. ln [acid] gave a slope of 0.28 indicating a fractional-order dependence of the rate on the H+ concentration (Fig. 2). In addition, the rate increased with increasing D2O content (data not shown). In order to indicate the nature of reactive species, the dielectric constant (D) of the solvent was varied by adding MeOH (5-25% v/v) to the reaction mixture, while the other experimental conditions were kept constant. Increased [MeOH] led to a rate enhancement (Table 2), and a plot of ln kobs vs. 1/D was linear (Fig. 3), (R2 = 0.976) with a positive slope. Values of D reported in the literature for MeOH-H2O mixtures were employed.[11] Data show that the rate of oxidation increase with a decrease in D (i.e., an increase in MeOH content) of the medium, which parallels the solution polarity. Control experiments indicated that MeOH was not oxidized by manganese(III) under the experimental conditions.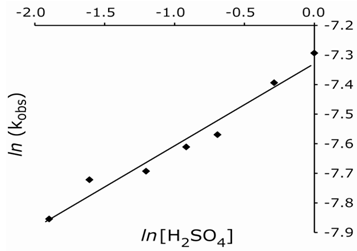 | Figure 2. Plot of ln kobs vs. ln [H2SO4] |
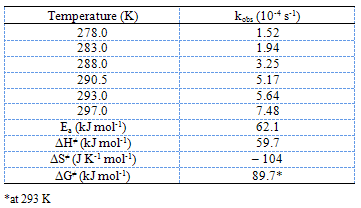
Finally, the oxidation reaction was studied at different temperatures, 278-297 K, keeping all other experimental conditions constant. Temperature dependent kobs values are given in Table 3 along with calculated activation parameters. From Arrhenius and Eyring plots of ln kobs vs.1/T and ln (kobs/T) vs.1/T, respectively, (Fig. 4) the energy of activation (Ea), entropy of activation (∆S≠), enthalpy of activation (∆H≠), and Gibbs free energy of activation (∆G≠) were computed.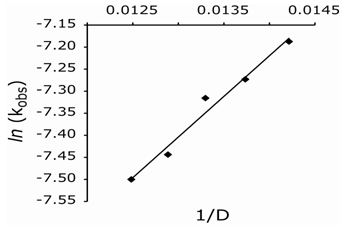 | Figure 3. Plot of ln kobs vs 1/D |
Table 3. Temperature dependent rates and activation parameters for the LA oxidation by manganese(III). [Mn(III)]o = 1.00 × 10-3 M; [LA]o = 2.00 10-2 M; [Mn(II)]o = 2.00 × 10-1 M; [H2SO4] = 3.50 10-1 M
 |
| |
|
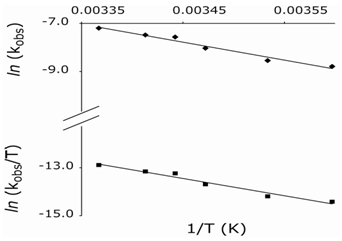 | Figure 4. Arrhenius (top) and Eyring plots |
3.1. Mechanism
Based on the kinetic results discussed above, a mechanism proposed for the LA-Mn(III) reaction in acid medium is as shown in Scheme II below.In Scheme II, the reactive oxidant species, Mn(III) sulfate complex anion, interacts with the substrate (LA) molecule in the fast pre-equilibrium (step (i)) to form an intermediate, substrate-oxidant complex anion, X. The complex anionic species, X, interacts with H+ in the rate-determining slow- step (ii) to form a nonionic complex intermediate, X1. Following the slow step, in successive fast steps, a mole of complex X1 interactions with three moles of Mn(III) sulfate species and a mole of H2O to form the end products, acetic acid, CO2 and Mn(II) sulfate, as indicated by product analysis and Scheme I.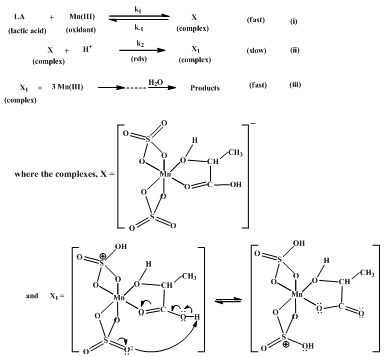 | Scheme II. Proposed mechanistic scheme for the lactic acid-Mn(III) reaction in acid medium |
3.2. Rate law derivation
From the rds or show step in Scheme II, 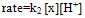 | (2) |
Application of the steady-state concept to the intermediate X in Scheme II leads to eq.(3).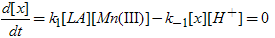 | (3) |
since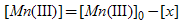 | (4) |
Substitution for [Mn(III)] from eq.(4). in eq.(3). and rearrangements for [x] lead to eq.(5).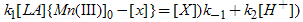 or
or 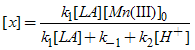 | (5) |
Substitution for [x] from eq.(5). into eq.(2). gives the rate law [eq.(6)].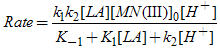 or
or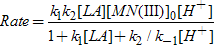 | (6) |
The rate law derived (eq. (6)) is in good agreement with the experimental kinetic results such as a first-order dependence of the rate on [Mn(III)] and a fractional order each on [LA] and [H+]. The proposed mechanism is also supported by the solvent effect. The dielectric constant effect (Fig. 3) shows the increased rate of oxidation with an increase in the MeOH content (or decreased D) of the solvent medium, which parallels the decreased solvent polarity. This effect is attributed to the formation of a relatively less polar transition state as compared to the ground state (slow step (ii), Scheme II). The less polar solvent mixture stabilizes the transition state and increases the rate[13,14]. The proposed mechanism is also supported by moderate values of energy of activation and other activation parameters. The fairly highly positive values of free energy of activation, enthalpy of activation and the negative entropy of activation (Table 3) indicate that a rigid, associative transition state formed is highly solvated[13,14]. Furthermore, the negative activation entropy suggests that the redox reaction is controlled by entropy rather than enthalpy.
4. Conclusions
A kinetic aspect of LA oxidation by Mn(III) in acid solutions has been studied spectrophotometrically at λmax = 491 nm at 20℃. Kinetic runs were performed under pseudo first order conditions of [LA] >> [Mn(III)]. The order of the reaction for each reactant was graphically determined to get the following experimental rate law: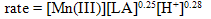 The reaction stoichiometry determined showed that four moles of Mn(III) were consumed per mole of LA in acid medium. The main reaction products, acetic acid, carbon dioxide, and Mn(II), were characterized. The activation parameters, such as Ea, ∆H≠, ∆S≠ and ∆G≠, were calculated to understand whether the reaction is controlled by entropy or enthalpy. The negative value of ∆S≠ suggests that the redox reaction is entropy controlled. A mechanism has been proposed for the LA oxidation, which supports the derived rate law that is consistent with the kinetic data.
The reaction stoichiometry determined showed that four moles of Mn(III) were consumed per mole of LA in acid medium. The main reaction products, acetic acid, carbon dioxide, and Mn(II), were characterized. The activation parameters, such as Ea, ∆H≠, ∆S≠ and ∆G≠, were calculated to understand whether the reaction is controlled by entropy or enthalpy. The negative value of ∆S≠ suggests that the redox reaction is entropy controlled. A mechanism has been proposed for the LA oxidation, which supports the derived rate law that is consistent with the kinetic data.
ACKNOWLEDGEMENTS
Authors are grateful to the Western Illinois University Research Council for financial support.
References
| [1] | Chidankumar, C. S., Chandraju, S. and Made Gowda, N. M. Synth. React. Inorg., Met.-Org., Nano-Met. Chem. 2009, 39, 645–650; Chandraju, S., Sherigara, B. S., Made Gowda, N.M, Int. J. Chem. Kinet., 1994, 26, 1105-1119; Sherigara, B.S., Ishwara Bhat, K., Pinto, I. and Made Gowda, N. M., ibid, 1995, 27, 675-690. |
| [2] | Seikichi, I. and Akira T. J. Biochem. 1954, 41, 187-198. |
| [3] | Samal, P. C., Pattnaik, B. B., Dharma Rao, S. Ch. and Mahapatro S. N. Tetrahedron, 1983, 39, 143-147. |
| [4] | Sapana, J., Hiran, B. L. and Bhatt, C. V. Electron. J. Chem. 2009, 6, 273-280. |
| [5] | Khan, Z., Raju, A. M. and -ud-Din, K. Int. J. Chem. Kinet. 2004, 36, 359-366. |
| [6] | Csavdari, A. and Baldea, I. Stud. Univ. Babes-Bol., Ser. Chem. 2007, LII, 35-45. |
| [7] | Ponnusamy, K., Shanmugakani, C., Sundaram, S. and Srinivasan, V. S. Inorg. Chem. 1987, 26, 968-970. |
| [8] | Abiraj, K., Ramesha Baba A. and Gowda, D. C. J. Phys. Org. Chem. 2006, 19, 68-74. |
| [9] | Bareka, J., Berka A. and Hlandikova, A. Micro. Chem. J. 1979, 24, 316-322. |
| [10] | Cholkar, K., Kouassi, G., Ananda, S., Veeraiah, M. K. and Made Gowda, N. M. Synth. React. Inorg., Met.-Org., Nano-Met. Chem. 2011, 41(9), 1126-1134. |
| [11] | Vogel A. I., Qualitative Organic Analysis (Longman and Green, London) (1958) p.708; Woolley, E. M., Tomkins, J. and Hepler, L.G. J. Solution Chem. 1972, 1, 341-351. |
| [12] | Waters, W A. and Littler, J.S., Oxidation in Organic Chemistry, Wiberg, K.B. ed., Academic Press, New York, 1965 pp.185-241. |
| [13] | Laidler K.J and Eyring H, Ann N Y Acad. Sci, 1940, 39 (1940) 303. |
| [14] | Collins, C. J. and Bowmann, N.S., Isotope Effects in Chemical Reactions , Van Nostrns New York, 1970, p.267. |
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTML