-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2012; 2(2): 26-31
doi: 10.5923/j.ajoc.20120202.05
Synthesis, Antibacterial and Antifungal Activities of Some Pyrazole-1-Carbothioamides and Pyrimidine-2(1H)-Thiones
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLEssam Mohamed Sharshira 1, Nagwa Mohamed Mahrous Hamada 2
1Department of Chemistry, Faculty of Science, Alexandria University, Alexandria .426, Egypt
2Department of Chemistry, Faculty of Education, Alexandria University, Alexandria, 21526, Egypt
Correspondence to: Nagwa Mohamed Mahrous Hamada , Department of Chemistry, Faculty of Education, Alexandria University, Alexandria, 21526, Egypt.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
A series of 3,5-disubstituted pyrazole-1-carbothioamides 2a-d, 4a-d, 5a-d and pyrimidine-2(1H)-thiones 6a-d were prepared by cyclocondensation of chalcones 1a-d with either thiosemicarbazide or thiourea, respectively. Dehydrative cyclization of chalcones 1a-d with thiosemicarbazide in refluxing dioxane containing few drops of acetic acid gave the corresponding 4,5-dihydropyrazoles 2a-d which were subsequently oxidized with sodium hypochlorite in dioxane to give pyrazole-1-carbothioamides 4a-d. N-acetyl derivatives of pyrazole-1-carbothioamides were obtained by two routes. Treatment of either hydrazones 3a-d or pyrazole-1-carbothioamides 4a-d with acetic anhydride gave the desired N-acetyl derivatives of pyrazole-1-carbothioamides 5a-d. Finally, cyclocondensation of chalcones 1a-d with thiourea in ethanolic potassium hydroxide gave the corresponding pyrimidine-2-(1H)-thiones 6a-d. The structural identification of products is reported and also the heterocyclic compounds were screened for their antimicrobial activity.
Keywords: chalcones, hydrazones, pyrazoles, pyrimidines
Article Outline
1. Introduction
- α,β-Unsaturated ketones display a wide range of pharmacological properties, including cytotoxity towards different cancer cell lines[1,2], antimitotic[3], antimutagenic[4], antibacterial[5], antiviral[6], anti-inflammatory[7] and hepatoprotective activities[8]. They are well known intermediates for synthesizing various heterocyclic compounds like pyrazoline and pyrimidine derivatives. A survey of literature in the recent past reveals that some pyrazoline derivatives possess antibacterial[9], anti-inflammatory[10] and antifungal effects[11]. Pyrimidine nucleus is an essential component in natural products like nucleic acids and vitamin B1. Also,different pyrimidine derivatives have remarkable pharmaceutical importance because of their biological activity as anti HIV, antitubercular and antidiabetic compounds[12,13]. In the view of the above mentioned facts and our continued interest in the synthesis of heterocyclic compounds derived from chalcone precursors[14-18], it was thought of interest to synthesize some new heterocyclic compounds containing pyrazoline and pyrimidine rings[19-26] and examination of their antimicrobial proper ties.
2. Results and Discussion
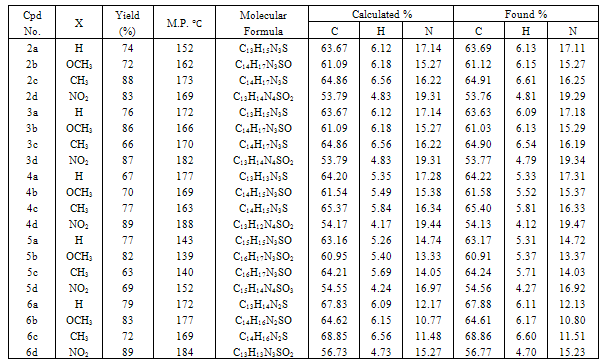
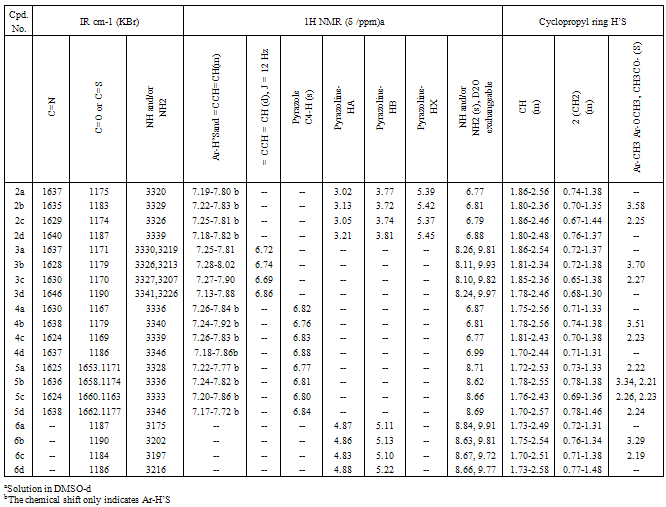
- Chalcones 1a-d for heterocyclic compounds synthesis were prepared in good yields by base catalyzed condensation of cyclopropylmethyl ketone with appropriately substituted benzaldehydes[16,27,28].The synthetic routes to our prepared compounds are shown in Scheme 1.Refluxing of chalcones 1a-d with thiosemicarbazide in dioxane containing few drops of acetic acid gave pyrazolin-1-carbothioamides 2a-d in good yields. The IR of 2 showed the presence of bands characteristic for a thioamide function at 1174-1187 (C=S) and 3320-3339 cm-1 (NH2). The pyrazoline ring CH2 protons appeared as a pair of doublets of doublets δ = 3.02-3.21 ppm and δ = 3.72-3.81 ppm. The CH proton (HX) appeared as a doublet of doublets at δ = 5.37-5.45 ppm due to vicinal coupling with the two magnetically non-equivalent protons of the methylene group (HA and HB) at position 4 of the pyrazoline ring (JAB = 16Hz, JAX = 3.6Hz, JBX = 12Hz). The cyclopropyl ring protons appeared as two multiplets in the range δ = 1.80-2.56 ppm (CH) and δ = 0.67-1.44 ppm (2CH2), respectively. When chalcones 1a-d were heated with thiosemicarbazide in ethanol containing few drops of acetic acid, thiosemicarbazones 3a-d were obtained in good yields. The structures of the prepared compounds were determined by spectral methods. The IR of the isolated thiosemicarbazones revealed characteristic bands for C=N at 1628-1646, C=S at 1170-1190, primary and secondary amines at 3326-3341 and 3207-3226 cm-1. The 1H NMR spectra revealed the presence of two broad exchangeable singlets at δ = 8.10-8.26 ppm and δ = 9.81-9.97 ppm characteristic for the NH2 and NH protons, respectively. The aromatic and =C-CH=CH protons appeared as a multiplet at δ = 7.13-8.02 ppm whereas the olefinic =C-CH=CH proton appears as a doublet in the range 6.69-6.86 ppm. The characteristic signals of cyclopropyl ring protons were also observed (Table 2). Oxidation of pyrazoline 2a-d using NaOCl in dioxane gave pyrazole-1- carbothioamides 4a-d in good yields. The IR of 4a-d showed the characteristic bands for C=N at 1624-1638 and thioamide function at 1167-1186 (C=S) and 3336-3346 cm-1 (NH2), while the 1H NMR spectra revealed a singlet at δ = 6.76-6.88 ppm for the pyrazole-C4-H. The N-acetyl derivatives 5a-d were obtained by two different methods. In the first method, pyrazoles 4a-d were heated under reflux with acetic anhydride, while in the second one, thiosemicarbazones 3a-d were cyclized to N-acetylpyrazoles 5a-d using acetic anhydride. The IR spectra of 5 showed the characteristic bands of carbonyl group at 1653-1662 cm-1. The 1H NMR of 5a-d exhibited a singlet of one proton intensity at δ = 6.77-6.84 ppm and another singlet of three protons intensity at δ = 2.21-2.24 ppm characteristic for pyrazole-C4-H and N-acetyl protons, respectively. Finally, when chalcones 1a-d were treated with thiourea in ethanolic potassium hydroxide solution, pyrimidine-2-thione derivatives 6a-d were obtained. The reaction occurred in two steps: first conjugate addition took place on the β-position of carbonyl group and then nucleophilic attack on the carbonyl group followed by dehydration led to the six-membered ring products[29,30] . The structure of 6 was indicated by spectral mehods, its IR spectrum reveals characteristic bands at 1184-1190 (C=S) and 3175-3216 cm-1 (NH). The 1H NMR spectrum of 6 showed the presence of two broad exchangeable singlets at δ = 8.63-8.84 ppm and δ = 9.72-9.91 ppm characteristic for two secondary amine protons whereas, the pyrimidine-C4-H and pyrimidine–C5-H appeared at δ = 4.83-4.88 ppm and δ = 5.10-5.22 ppm respectively. The structure of all newly isolated compounds was supported by measuring melting point and elemental analyses data showed in Table 1. All other characteristic spectra were indicated in Table 2.
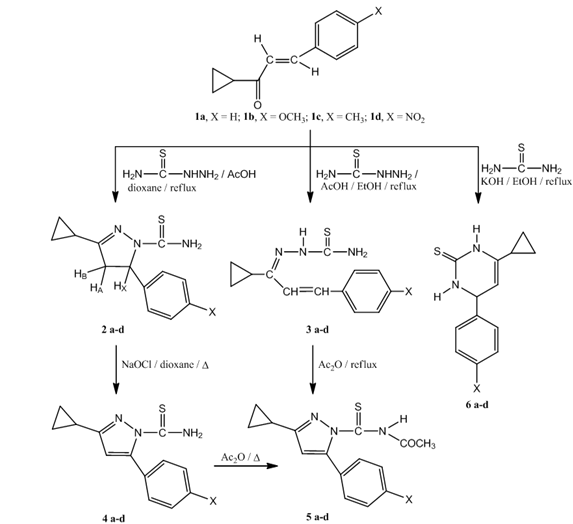 | Scheme 1. Synthesis of 2a-d, 3a-d, 4a-d, 5a-d and 6a-d |
|
|
|
2.1. Antimicrobial Activity
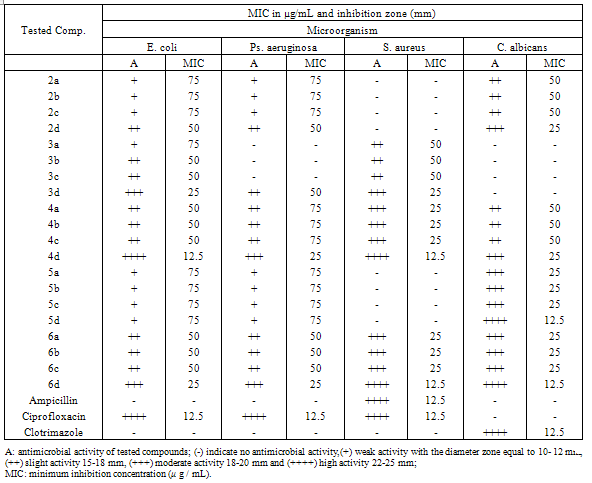
- The antimicrobial activities of the synthesized compounds were determined in vitro against several pathogenic representative microorganism Staphylococcus aureus ATCC6538P, Pseudomonas aeruginosa ATCC9027, Escherichia coli ATCC8739 and Candida albicans ATCC2091, using Agar well-diffusion method[31] . Ampicillin, Ciprofloxacin and Clotrimazole were used as standard drugs for studying the potential activities of these compounds. All the compounds were tested at different concentration level, DMSO was used as solvent and as control. The inhibition zone diameter in mm (IZD) was used as a criterion for the antimicrobial activity. The lowest concentration required to arrest the growth of bacteria was regarded as minimum inhibitory concentration (MIC, μg/mL), was determined for all the compounds and compared with the control. The investigation of antibacterial screening data revealed that carbothioamide derivatives 2a-d, 3a-d, 4a-d, 5a-d showed high activity against all the microorganisms employed in contrast with the carboxamide derivatives28, which exhibited no activity against Pseudomonas aeruginosa. on the other hand, compounds 6a-d exhibited interesting high biological activities with a degree of variation. The maximum activity (++++) (MIC = 12.5 μg/mL) was indicated for compound 4d, against Escherichia coli and Staphylococcus aureus and compound 6d against Staphylococcus aureus and Candida albicans. These results suggest that the electron-withdrawing NO2 group plays a crucial role in enhancing the observed activity. Moreover, compounds 4d exhibited a moderate activity (+++) (MIC = 25μg/mL) against Pseudomonas aeruginosa and Candida albicans whereas, 6d exhibited a moderate activity (+++) (MIC = 25μg/mL) against Escherichia coli and Pseudomonas aeruginosa. Compounds 3d, 4a, 4b, 4c, 4d, 6a, 6b, 6c and 6d were found to be the most active against all the microorganisms employed for both antibacterial and antifungal activity. In summary, all of our synthesized compounds showed a high and a moderate activity against Candida albicans and Pseudomonas aeruginosa. The results are summarized in Table 3.
3. Experimental
3.1. General
- Melting points were taken in open capillary tubes using Electrothermal apparatus 9100 (UK) and are uncorrected. Microanalyses were performed at Faculty of Science, Cairo University, Cairo, Egypt, using an Elementary Vario el III C, H, N, S Analyzer (Germany). IR spectra were recorded using potassium bromide disks on a Spectrum RXI/FT-IR System Perkin Elmer, USA (Faculty of Pharmacy, Alexandria University, Alexandria, Egypt). 1H NMR spectra were determined on a Varian EM-390 MHz spectrophotometer, using TMS as internal standard.
3.2. General Procedure for Preparation of E-l-Cyclopro- pyl-3-(p-substituted-phenyl)-2-propen ones 1a-d
- Compounds 1a-d were obtained in a good yields according to published method[27]. The physical properties and all the spectral data were as reported in the literature.
3.3. General Procedure for Preparation of 4,5-Dihydro-3 -cyclopropyl-5-(p-substituted-phenyl)-pyrazole-1- carbothioamides 2a-d
- A solution of chalcones 1a-d (10 mmol) in dioxane (10 mL) was refluxed with the appropriate thiosemicarbazide (10 mmol) in glacial acetic acid (1 mL) for 4 hours, then the reaction mixture was poured onto crushed ice and was kept overnight at room temperature, the separated solid was filtered off, washed successively with water and dried, then recrystallized from methanol. Melting points, IR and NMR data: see Tables 1 and 2.
3.4. General Procedure for Preparation of 1-Cyclopropyl -3-(p-substituted-phenyl)-2-propene-1-thiosemicarbazones 3a-d
- A solution of chalcones 1a-d (10 mmol) in ethanol (10 mL) was refluxed with the appropriate thiosemicarbazide (10 mmol) in glacial acetic acid (2 mL) for about five hours, then the reaction mixture was treated as mentioned for 2a-d. Melting points and spectral data are listed in Tables 1 and 2.
3.5. General Procedure for Preparation of 3-Cyclopropyl -5-(p-substituted–phenyl)-pyrazole-1- carbothioamides 4a-d
- A solution of the appropriate pyrazoline 2a-d (10 mmol), dioxane (10 mL) and sodium hypochlorite (5 mL, 5%) was heated over a boiling water bath until effervescence occurs: heating was continued for a further 10 minutes. The reaction mixture was allowed to reach ambient temperature and the separated solid was filtered, washed with water, dried and recrystallized from methanol to give the corresponding pyrazoles 4a-d. Melting points, IR and NMR data: see Tables 1 and 2.
3.6. General Procedure for Preparation of 3-Cyclopropyl -5-(p-substituted-phenyl)-pyrazole-1- (N-acetyl) carboth- ioamides 5a-d
- Method A: a mixture of the appropriate thiosemicarbazone 3a-d (10 mmol) and acetic anhydride (15 mL) was heated under reflux for three hours. After the reaction mixture attained room temperature, it was poured into crushed ice and the oily product deposited was decanted from water and extracted with ether. The ether layer was washed with sodium bicarbonate, followed by water, dried over anhydrous sodium sulphate and evaporated to give the corresponding pyrazoles 5a-d as needles. Melting points, elemental analyses, IR and NMR data: see Tables 1 and 2.Method B: A mixture of pyrazoles 4a-d (10 mmol) in acetic anhydride (5 mL) was heated under reflux for 30 minutes. The reaction mixture was treated as mentioned in method A to give the N-acetyl derivatives 5a-d.
3.7. General Procedure for Preparation of 3,4-Dihydro-6 -cyclopropyl-4-(p-substituted-phenyl)-pyrimidin-2(1H)- thione 6a-d
- A mixture of chalcone (10 mmol), thiourea (10 mmol) and potassium hydroxide (1.0 g) in ethanol (40 ml) was refluxed on a boiling water bath for one hour. The reaction mixture was left overnight and then concentrated under reduced pressure. The solid residue was collected, washed with water and recrystallized from ethanol. Melting points, IR and NMR data: see Tables 1 and 2.
3.8. Determination of Antimicrobial Activity
- Cultures of four different microorganisms namely: Escherichia coli (E. coli), Pseudomonas aeruginosa (Ps. aeruginosa), Staphylococcus aureus (S. aureus), Candida albicans (C. albicans) were used to investigate the antimicrobial activities of compounds 3a-d, 4a-d, 5a-d and 6a-d. The antimicrobial activities were assayed biologically using diffusion plate technique. The experiments were carried out by pouring a spore suspension 10 6 colon-forming units (CFU) per mL of the test strain to 75 mL of nutrient agar medium at 45℃ mixed well, and then poured into a 15 cm sterile metallic Petri plate. The medium was allowed to solidify and 8 mm wells were dug with a sterile metallic borer, then, a DMSO solution of the test sample (1 mL) at 1 mg/mL was added to the respective wells. DMSO was served as negative control and the standard antimicrobial drugs Ampicillin, Ciprofloxacin and Clotrimazole were used as positive controls. The layer was allowed to set for 30 minutes and incubated at optimum incubation temperature 28 ± 2℃. Test organism growth may be affected by the inhibitory action of the test compound and so, a clear zone around the disc appeared as an indication of the inhibition of the test organism growth. The size of clearing zone is proportional to the inhibitory action of the test compound. Measurements were considered after 72 h for fungi and 24 h for bacteria. The results are shown in Table 3.
4. Conclusions
- This work describes different methods for the synthesis of new heterocyclic compounds such as pyrimidine and pyrazole derivatives. The antimicrobial activities of these compounds were evaluated against Gram-positive, gram- negative bacteria and fungi. Most of the synthesized compounds showed a moderate antimicrobial activity.