-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2012; 2(2): 7-13
doi: 10.5923/j.ajoc.20120202.02
Utility of Enaminonitriles in Heterocyclic Synthesis: Synthesis and Antimicrobial Activity of Some New Azole and Azine Derivatives
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLAhmed A. Fadda 1, Adel A.-H. Abdel-Rahman 2, Ezzat A. Hamed 3, Ekbal H. Khalil 2
1Chemistry Department, Faculty of Science, Mansoura University, 35516 Mansoura, Egypt
2Chemistry Department, Faculty of Science, Menoufia University, Menoufia, Egypt
3Chemistry Department, Faculty of Science, Alexandria University, Alexandria, Egypt
Correspondence to: Ahmed A. Fadda , Chemistry Department, Faculty of Science, Mansoura University, 35516 Mansoura, Egypt.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Reactions of 6-amino-3-methyl-1,4-diphenyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (1) with a variety of reagents leads to the synthesis of pyrano[2,3-c]pyrazole derivatives has been investigated with the aim to explore the use of this exceptionally reactive nitrile in heterocyclic synthesis.
Keywords: Dihydropyrano[2,3-c]pyrazole, Enaminonitriles, Azoles, Pyrimidines, Pyridines
Article Outline
1. Introduction
- Previous papers have shown that pyran derivatives possess pronounced chemical and biological properties[1-3]. On the other hand, substituted pyridines show acaricidal, insecticidal and herbicidal activities [4]. Moreover, pyrimidines are important analgesic and anti-inflammatory agents [5, 6]. Compounds having a combination of cyclohexylpyran with pyridine and/or pyrimidine moieties can be expected to possess medicinal properties. In addition, some compounds of this class, notably 3-phenylcoumarin containing an azole ring have found applications as fluorescent brightening agent [7]. In contrast to most other types of tumer inhibitory compounds, many of which exhibit toxicity, mutagenicity and other undesirable properties, the pyranopyridine and pyranopyrimidine compounds tend to show minimal side effects. The formation of a new fused heterocyclic ring is an important task for heterocyclic chemists from various points of view.Also, o-aminonitriles and their versatile role as synthetic intermediates are ideally suited since they consist of multifunctional building unite for new and promising compounds in one or two easy reaction steps. In the last few years Fadda et al., has been involved in an exploration of the potential of activated nitriles in heterocyclic synthesis [8-12]. From these above facts and as part of our program, the reactivity of 6-amino-3-methyl-1,4-diphenyl-1,4-dihydropyrano[2,3-c] pyrazole-5-carbonitrile (1) towards a variety of reagents has been investigated with the aim to explore the use of this exceptionally reactive nitrile in heterocyclic synthesis.
2. Results and Discussion
2.1. Chemistry
- The synthetic procedures adopted to obtain the target compounds are depicted in Schemes 1-3. The starting 6-amino-3-methyl-1,4-diphenyl-1,4-dihydropyrano-[2,3-c]pyrazole-5-carbonitrile (1) was prepared according to the previously reported methods[13,14]. It is well known that, activation of cyanoacetic acid by conversion to the mixed anhydride with acetic anhydride has been used[15-18] but the generality, simplicity and usefulness has not been appreciated and the reagent has infrequently been used for N-acetylations of e.g. urea and C-acetylation of enamines [19]. Other activation procedures, such as conversion to cyanoacetyl chloride have also been used, albeit this reagent is notorious for its tendency to self polymerization, particularly when heated[20]. However, heating of cyanoacetic acid together with dihydropyrano[2,3-c]pyrazole derivative acetic anhydride gave the desired product in an excellent yields of acetamide derivative 2 as a readily collectable precipitate. Thus, it was found that refluxing of ethanolic solution of 1 with hydrazine hydrate in presence of a catalytic amount of piperidine yielded the corresponding 3-amino-1,4,7-trihydro-5-methyl-4,7-diphenyl-pyrazolo[3', 4',2,3]pyrano[6,5-c]pyrazole (3). Its IR spectrum displayed absorption bands at 3429, 3264 and 3095 cm-1 due to NH2 and NH groups and showed no absorption at the CN region. Also, the structure of 3 was judged by mass spectrum, it showed the molecular ion peak at m/z 344 (M++1, 41.2) which is in agreement with its molecular formula (C20H17N5O). The reaction proposed to proceed by addition of hydrazine molecule to the cyano group which followed by loss of ammonia molecule during refluxing for prolonged time with the formation of the isolable product 3. Similarly, heating an equimolar mixture of 1 and o- phenylenediamine, o-aminophenol or o-aminothiophenol in absolute ethanol in the presence of catalytic amount of piperidine for long time afforded the corresponding 5- (1H-benzo[d](imidazol/ oxazol /or thiazole)-2-yl)-3-methyl- 1,4-diphenyl-1,4-dihydro-pyrano[2,3-c]pyrazol-6-amine de- rivatives 4, 5 and 6, respectively. The reaction proceeds by initial addition of hydrogen to cyano group, which then undergoes intramolecular cyclization via loss of NH3 molecule which led to formation of the final products 4, 5 and 6, respectively (Scheme 1). Structures 4, 5 and 6 were established by the correct analyses and compatible spectroscopic data. In general, the IR spectra showed an absorption band at 3388-3190 cm-1 due to the stretching frequency of the NH2 group and the disappearance of CN group while in the 1H-NMR spectra of compounds 4 and 5 the NH2 protons appeared at δ 8.18 and 7.54 ppm, respectively as singlet signals. In addition, the mass spectroscopic measurement for compounds 4, 5 and 6 showed the molecular ion peaks at m/z 418 (M+-1, 12.5), 420 (M+, 13.3) and 421 (M+-CH3, 17.6), respectively.
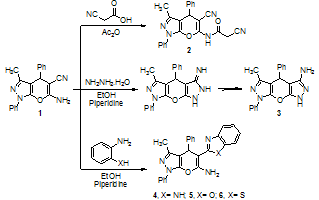 | Scheme 1 |
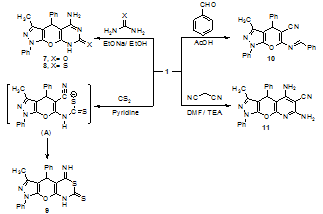 | Scheme 2 |
2.2. Antimicrobial Activity
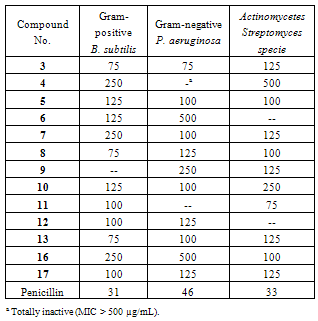
- The antimicrobial activity of the synthesized compounds was evaluated against three microorganisms; Bacillus subtilis (ATCC 6633) (Gram-positive), Pseudomonas aeruginosa (ATCC 27853) (Gram-negative) and Streptomyces species (Actinomycetes). The values of minimal inhibitory concentrations (MICs) of the tested compounds are presented in Table 1. The results of the antimicrobial activity test revealed that 3, 8, and 13 showed the highest activity against B. subtilis with MIC values of 75 µg/mL followed by compounds 11, 12, and 17. Compound 3 showed the highest inhibition activity against P. aeruginosa, whereas compound 11 was the most active among the series of tested compounds against Streptomyces species with MIC values of 75 µg/mL. The results also revealed that some compounds showed little or no activity against the microorganisms (Table 1).
|
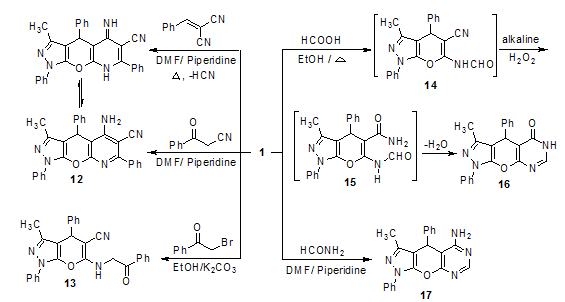 | Scheme 3 |
3. Experimental
- All melting points are recorded on Gallenkamp electric melting point apparatus. The IR spectra υ cm-1 (KBr) were on Perkin Elmer Infrared Spectrophotometer Model 157, Grating. The 1H-NMR spectra were run on Varian Spectrophotometer at 400 MHz using TMS as an internal reference and DMSO-d6 as solvent. The mass spectra (EI) were run at 70 eV with JEOL JMS600 equipment and/or a Varian MAT Spectrometer. Elemental analyses (C, H and N) were carried out at the Microanalytical Center of Cairo University, Giza, Egypt. The results were found to be in good agreement (±0.3%) with the calculated values. 6-Amino-3-methyl-1,4- diphenyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (1) was prepared according to the previously reported methods [13, 14] as white needles, mp 179-180oC; 79%; IR (KBr): ν/cm-1= 3470, 3322 (NH2), 2197 (CN), 1659 (C=N), 1592 (C=C), 1491 (Ph); MS: m/z (%): 328 (M+, 13.12), 262 (0.22), 251 (83.5), 185 (93.8), 174 (20.3), 154 (17.0), 127 (23.8), 91 (20.0), 77 (46.2), 66 (21.9).Synthesis of 2-cyano-N-(5-cyano-3-methyl-1,4-diphen yl-1,4-dihydropyrano[2,3-c]pyrazol-6-yl)acetamide (2). A solution of cyanoacetic acid (5 mmol) in acetic anhydride (15 mL) was heated under reflux over water bath for 5 minutes and dihydropyrano[2,3-c]pyrazole derivative 1 (5 mmol) was added. The reaction mixture was refluxed for further 1 h at 60-70℃, and then left to cool. The precipitated solid was filtered off, dried and recrystallized from ethanol to give acetamide derivative 2. Yield (54%); brown powder; mp 125℃; IR (KBr): ν/cm-1= 3395 (NH), 2926 (CH, aliphatic), 2192 (CN), 1598 (CO, amidic), 1498 (Ph); 1H-NMR (200 MHz, CDCl3) δ (ppm): 2.15 (s, 3H, CH3), 2.57 (s, 2H, CH2), 4.79 (s, 1H, CH), 7.26-7.96 (m, 10H, Ar-H), 8.46 (s, 1H, NH). MS (EI, 70 ev) m/z (%) = 395 (M+, 33.3), 394 (M+-1, 27.8), 266 (33.3), 233 (33.3), 226 (33.3), 210 (33.3), 211 (33.3), 91 (33.3), 87 (38.9), 68 (55.6), 50 (100.0). Anal. Calcd. for C23H17N5O2 (395.41): C, 69.86; H, 4.33; N, 17.71%. Found: C, 69.93; H, 4.37; N, 17.77%.Synthesis of 3-amino-1,4,7-trihydro-5-methyl-4,7- diphenyl-pyrazolo[3',4',2,3]pyrano[6,5-c]pyrazole (3). Equimolar amounts of (1, 5 mmol) and hydrazine hydrate (5 mol) in absolute ethanol (30 mL) in presence of a catalytic amount of piperidine (4 drops) was refluxed for 12 h. The reaction mixture was left to cool at room temperature and then poured in to cold water for complete precipitation. The solid products was filtered off and recrystallized from aqueous ethanol to give the corresponding compound 3. Yield (30.9%); black crystals; mp 160-162℃; IR (KBr): ν/cm-1= 3429, 3264 (NH2), 3095 (NH), 1595 (C=N), 1497 (Ph). MS (EI, 70 ev) m/z (%) = 344 (M++1, 41.2), 181 (35.5), 120 (23.5), 103 (35.5), 82 (41.2), 70 (35.3), 64 (100.0), 53 (41.2). Anal. Calcd. for C20H17N5O (343.38): C, 69.96; H, 4.99; N, 20.40%. Found: C, 70.04; H, 5.06; N, 20.47%.Reaction of dihydropyrano[2,3-c]pyrazole derivative 1 with o-substituted anilinesGeneral procedure: An equimolar amounts of (1, 5 mmol), o-phenylenediamine, o-aminophenol or o-aminothiophenol in absolute ethanol (30 mL) in the presence of a catalytic amount of piperidine (4 drops) was refluxed for 30 h. The reaction mixture was concentrated to its half volume and then left to cool at room temperature overnight. The solid products was filtered off and recrystallized from ethanol to give the corresponding dihydropyrano[2,3-c]pyrazole derivatives 4, 5 and 6, respectively.5-(1H-Benzo[d]imidazol-2-yl)-3-methyl-1,4-diphenyl-1,4-dihydropyrano[2,3-c]pyrazol-6-amine (4). Yield (35.4%); brown powder; mp 121-125℃; IR (KBr): ν/cm-1= 3383, 3360 (NH2), 3179 (NH), 1658 (C=N), 1499 (Ph); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 2.28 (s, 3H, CH3), 4.87 (s, 1H, CH), 7.24-7.81 (m, 14H, Ar-H), 8.18 (s, 2H, NH2), 8.32 (s, 1H, NH). MS (EI, 70 ev) m/z (%) = 418 (M+-1, 12.5), 334 (12.5), 200 (12.5), 186 (16.7), 174 (22.9), 91 (100.0), 50 (29.2). Anal. Calcd. for C26H21N5O (419.48): C, 74.44; H, 5.05; N, 16.70%. Found: C, 74.36; H, 5.01; N, 16.63%.5-(Benzo[d]oxazol-2-yl)-3-methyl-1,4-diphenyl-1,4-di-hydropyrano[2,3-c]pyrazol-6-amine (5). Yield (92.4%); pale grey powder; mp 158℃; IR (KBr): ν/cm-1= 3388, 3192 (NH2), 1594 (C-O), 1497 (Ph); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 2.37 (s, 3H, CH3), 3.75 (s, 1H, CH), 7.16-7.46 (m, 14H, Ar-H), 7.54 (s, 2H, NH2). MS (EI, 70 ev) m/z (%) = 420 (M+, 13.3), 302 (42.2), 262 (15.6), 222 (13.3), 185 (22.2), 152 (15.6), 93 (20.0), 77 (100.0). Anal. Calcd. for C26H20N4O2 (420.46): C, 74.27; H, 4.79; N, 13.33%. Found: C, 74.34; H, 4.82; N, 13.41%.5-(Benzo[d]thiazol-2-yl)-3-methyl-1,4-diphenyl-1,4-di-hydropyrano[2,3-c]pyrazol-6-amine (6). Yield (94%); yellowish green powder; mp 120-122℃; IR (KBr): ν/cm-1= : 3387, 3190 (NH2), 1596 (C=N), 1497 (Ph); MS (EI, 70 ev) m/z (%) = 421 (M+-CH3, 17.6), 358 (14.7), 269 (26.5), 268 (100.0), 267 (26.5), 241 (20.6), 211 (38.2), 179 (29.4), 162 (26.5), 134 (47.1), 108 (61.8), 91 (88.2), 69 (70.6). Anal. Calcd. for C26H20N4OS (436.53): C, 71.54; H, 4.62; N, 12.83%. Found: C, 71.59; H, 4.67; N, 12.91%.Synthesis of 5-amino-4,8,9-trihydro-3-methyl-1,4-di phenyl-pyrazolo[3',4',2,3]pyrano[6,5-d]pyrimidine-7-one (thione) derivatives 7 and 8General procedure: A mixture of (1, 5 mmol) and urea (5 mmol) or thiourea (5 mmol) in absolute ethanol (20 mL) and sodium ethoxide ( sodium metal in (20 mL) absolute ethanol, 5 mmol) was refluxed for 6 h. The reaction mixture was left to cool at room temperature, then poured in to ice cold water (50 mL) and neutralized with dilute hydrochloric acid. The separated material was filtered off and recrystallized from ethanol to yield compounds 7 and 8, respectively.5-Amino-4,8,9-trihydro-3-methyl-1,4-diphenyl-pyrazolo[3',4',2,3]pyrano[6,5-d]pyrimi-dine-7-one (7). Yield (75.2%); pale orange powder; mp 210oC; IR (KBr): ν/cm-1= 3067 (NH), 1654 (C=O), 1497 (Ph); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 1.57 (s, 3H, CH3), 4.48 (s, 1H, CH), 5.61 (s, 1H, NH2), 7.25-7.53 (m, 10H, Ar-H), 7.54 (s, 2H, NH). MS (EI, 70 ev) m/z (%) = 371 (M+, 0.29), 262 (89.86), 185 (100.0), 174 (62.89), 128 (35.85), 105 (14.01), 91 (30.79), 77 (64.58). Anal. Calcd. for C21H17N5O2 (371.39): C, 67.91; H, 4.61; N, 18.86%. Found: C, 67.96; H, 4.65; N, 18.94%.5-Amino-4,8,9-trihydro-3-methyl-1,4-diphenyl-pyrazolo[3',4',2,3]pyrano[6,5-d]pyrimi-dine-7-thione (8). Yield (34.4%); brown powder; mp 138-140oC; IR (KBr): ν/cm-1= 3323, 3217 (NH2), 1625 (C=N), 1482 (Ph), 1188 (C=S); 1H-NMR (200 MHz, CDCl3) δ (ppm): 2.04 (s, 3H, CH3), 4.53 (s, 1H, CH), 4.78 (s, 1H, NH), 5.65 (s, 2H, NH2), 7.11-7.60 (m, 10H, Ar-H), 7.54 (s, 2H, NH). MS (EI, 70 ev) m/z (%) = 372 (M+-CH3, 58.3), 359 (58.3), 358 (50.0), 345 (58.3), 298 (58.3), 245 (58.3), 192 (33.3), 174 (25.0), 156 (91.7), 115 (66.7), 78 (100.0), 52 (83.3). Anal. Calcd. for C21H17N5OS (387.46): C, 65.10; H, 4.42; N, 18.08%. Found: C, 65.18; H, 4.49; N, 18.13%.Synthesis of 4-imino-1,5,9-trihydro-6-methyl-4,7- diphenyl-pyrazolo[3',4',2,3]pyrano[6,5-d]thiazine-2-thione (9). To a solution of (1, 5 mmol) in dry pyridine (30 mL) was added carbon disulphide (5 mmol). The reaction mixture was refluxed on water bath for 6 h, then left to cool at room temperature, poured in to cold water and neutralized with dilute hydrochloric acid for complete precipitation. The obtainable solid was filtered off, washed with water, dried well, and recrystallized from methanol to yield compound 9. Yield (44.7%); light yellow powder; mp 140-144oC; IR (KBr): ν/cm-1= 3062 (2NH), 1595 (C=N), 1497 (Ph), 1180 (C=S), 757 (C-S). MS (EI, 70 ev) m/z (%) = 405 (M++1, 1.58), 358 (3.68), 346 (11.23), 314 (12.46), 276 (3.10), 262 (31.17), 185 (22.64), 174 (23.88), 128 (16.29), 105 (16.97), 91 (35.33), 77 (100.0). Anal. Calcd. for C21H16N4OS2 (404.51): C, 62.35; H, 3.99; N, 13.85%. Found: C, 62.42; H, 4.06; N, 13.94%.Synthesis of 6-(benzylideneamino)-3-methyl-1,4- diphenyl-1,4-dihydro-pyrano[2,3-c]pyrazole-5-carbonitrile (10). A mixture of (1, 5 mmol) and benzaldehyde (5 mmol) in glacial acetic acid (30 mL) was heated under reflux for 1 h, cooled, and poured into crushed ice. The produced solid was filtered off, washed with water, dried well, and recrystallized from a mixture of benzene and petroleum ether (3:1) to yield the corresponding compound 10. Yield (74%); white sheets; mp 220℃; IR (KBr): ν/cm-1= 2100 (CN), 1596 (C=N), 1500 (Ph), 1188 (C=S); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 2.17 (s, 3H, CH3), 5.04 (s, 1H, CH), 6.11 (s, 1H, N=CH), 7.22-7.28 (m, 15H, Ar-H). MS (EI, 70 ev) m/z (%) = 339 (M+-Ph, 0.1), 304 (58.99), 286 (8.67), 262 (100.0), 200 (1.89), 185 (38.1), 174 (19.26), 128 (18.9), 106 (35.88), 91 (10.2), 77 (42.74). Anal. Calcd. for C27H20N4O (416.47): C, 77.87; H, 4.84; N, 13.45%. Found: C, 77.95; H, 4.94; N, 13.57%.Synthesis of 5,7-diamino-6-cyano-4,9-dihydro-3- methyl-1,4-diphenyl-pyrazolo[3',4',2,3]pyrano[6,5-b]-pyridine (11). To a solution of (1, 5 mmol) in DMF (30 mL) was added malononitrile (5 mmol) followed by few drops of TEA (4 drops). The reaction mixture was reflux for 6 h, left to cool at room temperature overnight and then poured in to cold water (50 mL). The obtainable solid was filtered off, washed with water, dried well, and recrystallized from ethanol to give compound 11. Yield (98.2%); yellowish green powder; mp 120℃; IR (KBr): ν/cm-1= 3128, 3026 (NH2), 2193 (CN), 1601 (C-O), 1499 (C=N), 1499 (Ph); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 2.47 (s, 3H, CH3), 3.94 (s, 1H, CH), 6.94 (s, 2H, NH2), 7.14 (s, 2H, NH2), 7.38-7.48 (m, 10H, Ar-H). MS (EI, 70 ev) m/z (%) = 350 [M+-2-(N=C-NH2), 1.0], 284 (100.0), 257 (16.1), 230 (7.95), 219 (8.3), 203 (6.42), 165 (20.71), 138 (5.11), 115 (2.22), 77 (7.33), 66 (5.45). Anal. Calcd. for C23H18N6O (394.43): C, 70.04; H, 4.60; N, 21.31%. Found: C, 70.11; H, 4.67; N, 21.38%.Synthesis of 5-amino-6-cyano-4,9-dihydro-3-methyl- 1,4,7-triphenyl-pyrazolo [3',4',2,3]pyrano[6,5-b]pyridine (12). Method A. To a solution of (1, 5 mmol) in DMF (30 mL) was added phenacylcyanide (5 mmol) followed by a few drops of piperidine. The reaction mixture was reflux for 6 h, cold, poured in to ice-cold water for complete precipitation and neutralized by dil. HCl for complete precipitation. The precipitated solid was collected by filtration, and recrystallized from aqueous ethanol to yield compound 12. Yield (42.7%); brown powder; mp 135℃; IR (KBr): ν/cm-1= 3134, 3039 (NH2), 2183 (CN), 1596 (C-O), 1497 (Ph). MS (EI, 70 ev) m/z (%) = 454 (M+-1, 4.9), 453 (M+-2, 15.72), 452 (M+-3, 14.88), 399 (5.14), 382 (5.59), 301 (12.6), 262 (76.7), 185 (64.3), 174 (39.2), 128 (27.1), 105 (21.4), 91 (81.8), 77 (100.0). Anal. Calcd. for C29H21N5O (455.51): C, 76.47; H, 4.65; N, 15.37%. Found: C, 76.52; H, 4.71; N, 15.42%.Method B. An equimolar amounts of (1, 5 mmol) and benzylidine malononitrile (5 mmol) was refluxed for 6 h in a mixture of DMF in the presence of few drops of piperidine (4 drops). The reaction mixture was left to cool, poured into ice-cold water for complete precipitation, then filtered off and recrystallized from aqueous ethanol to yield compound 12.Synthesis of 3-methyl-6-(2-oxo-2-phenylethylamino)- 1,4-diphenyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (13). A mixture of (1, 5 mmol) and phenacyl bromide (5 mmol) in absolute ethanol (30 mL) in presence of anhydrous potassium carbonate (5 mmol) as a catalyst was refluxed for 3 h. The reaction mixture was left to cool at room temperature overnight. The separated material was filtered off and recrystallized from ethanol to yield compound 13. Yield (43.4%); brown powder; mp 126-128℃; IR (KBr): ν/cm-1= 3657 (NH), 2196 (CN), 1661 (CO), 1597 (C=N), 1494 (Ph); 1H-NMR (200 MHz, CDCl3) δ (ppm): 1.95 (s, 3H, CH3), 3.8 (s, 2H, CH2), 5.48 (s, 1H, CH), 7.24-7.52 (m, 15H, Ar-H), 8.29 (s, 1H, NH). MS (EI, 70 ev) m/z (%) = 420 (M+-CN, 2.87), 418 (4.3), 394 (2.88), 370 (3.5), 360 (3.17), 314 (6.36), 293 (10.9), 275 (6.95), 262 (13.7), 200 (5.99), 185 (14.0), 174 (9.56), 156 (4.01), 128 (11.3), 105 (76.7), 91 (38.6), 77 (100.0). Anal. Calcd. for C28H22N4O2 (446.5): C, 75.32; H, 4.97; N, 12.55%. Found: C, 75.38; H, 5.06; N, 12.67%.Synthesis of 4,6,9-trihydro-3-methyl-1,4-diphenyl- pyrazolo[3',4',2,3]pyrano[6,5-d]pyrimidine-5-one (16). An equimolar amounts of (1, 5 mmol) and formic acid (5 mmol) in absolute ethanol (30 mL) was refluxed for 2 h. Potassium carbonate (10%, 10 mL) and hydrogen peroxide (5 mL) were added to the reaction mixture and continued refluxing for further one hour. The reaction mixture was concentrated and left to cool at room temperature overnight for complete precipitation. The separated solid was collected by filtration, and recrystallized from aqueous ethanol to yield compound 16. Yield (42.2%); brown crystals; mp 120℃; IR (KBr): ν/cm-1= 3061 (NH), 1659 (CO, amidic), 1599 (C=N), 1496 (Ph); 1H-NMR (200 MHz, CDCl3) δ (ppm): 1.96 (s, 3H, CH3), 4.79 (s, 1H, CH-Ph), 7.18-7.36 (m, 10H, Ar-H), 7.63 (s, 1H, -CH=N), 10.02 (s, 1H, NH). MS (EI, 70 ev) m/z (%) = 358 (M++2, 0.52), 357 (M++1, 0.2), 346 (1.7), 320 (6.5), 304 (10.8), 262 (100.0), 185 (43.7), 174 (42.3), 128 (18.7), 107 (16.2), 91 (4.2), 77 (12.4). Anal. Calcd. for C21H16N4O2 (356.38): C, 70.77; H, 4.53; N, 15.72%. Found: C, 70.83; H, 4.61; N, 15.77%.Synthesis of 5-amino-4,9-dihydro-3-methyl-1,4- diphenyl pyrazolo[3',4',2,3]pyrano[6,5-d]pyrimidine (17). Equimolar amounts of (1, 5 mmol) and formamid (5 mmol) in dimethylformamide (30 mL) followed by few drops of piperidine the reaction mixture was refluxed for 6 h, then cold and poured into ice cold water and neutralized by dilute hydrochloric acid for complete precipitation. The precipitated solid was filtered off, dried and recrystallized from aqueous ethanol to give 17. Yield (62.7%); pale brown powder; mp 100oC; IR (KBr): ν/cm-1= 3059, 3032 (NH2), 1598 (C=N), 1496 (Ph). MS (EI, 70 ev) m/z (%) = 340 (M+-CH3, 6.49), 262 (100.0), 261 (38.6), 185 (75.3), 174 (41.5), 128 (33.1), 105 (13.7). Anal. Calcd. for C21H17N5O (355.39): C, 70.97; H, 4.82; N, 19.71%. Found: C, 71.09; H, 4.93; N, 19.76%.Sample preparationEach of the test compounds and standards were dissolved in 12.5% DMSO, at concentrations of (500 µg/mL). Further dilutions of the compounds and standards in the test medium were prepared at the required quantities.Culture of microorganismsBacteria strains were supplied from Botany Department, Faculty of Science, Menoufia University, Shebin El-Koam, Egypt, namely Bacillus subtilis (ATCC 6633) (Gram- positive), Pseudomonas aeruginosa (ATCC 27853) (Gram- negative) and Streptomyces species (Actinomycetes). The bacterial strains were maintained on MHA (Mueller – Hinton agar) medium (Oxoid, Chemical Co., UK) for 24 h at 37℃. The medium was molten on a water bath, inoculated with 0.5 mL of the culture of the specific microorganism and poured into sterile Petri dishes to form a layer of about 3- thickness. The layer was allowed to cool and harden. With the aid of cork-borer, cups of about diameter were produced[28].Agar diffusion technique The antibacterial activities of the synthesized compounds were tested against Bacillus subtilis (Gram-positive), Pseudomonas aeruginosa (Gram-negative) and Streptomyces species (Actinomycetes) using MH medium ( casein hydrolysate, soluble starch, 1000 mL beef extract). A stock solution of each synthesized compound (500 µg/mL) in DMSO was prepared and graded quantities of the test compounds were incorporated in specified quantity of sterilized liquid MH medium. Different concentrations of the test compounds in DMF were placed separately in cups in the agar medium. All plates were incubated at 37℃ overnight. The inhibition zones were measured after 24 h. The minimum inhibitory concentration (MIC) was defined as the intercept of the grave of logarithm concentrations versus diameter of the inhibition zones[29,30].