-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2012; 2(1): 39-47
doi: 10.5923/j.ajoc.20120201.08
Heteroannulation of Pyrido[2,3-d]Pyrimidines. Synthesis and Spectral Characterization of Pyridotriazolopyrimidines, Pyridopyrimidotriazine and Pyridopyrimidotriazepine Derivatives
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLMahmoud R. Mahmoud , Hassan M. F. Madkour , Mohamed M. Habashy , Amr M. El-Shwaf
Chemistry Department, Faculty of Science, Ain Shams University, Abbassia, Cairo, 11566, Egypt
Correspondence to: Mahmoud R. Mahmoud , Chemistry Department, Faculty of Science, Ain Shams University, Abbassia, Cairo, 11566, Egypt.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Number of pyridotriazolo-, pyridothiazolo-, pyridotetrazolopyrimidines and pyrido-pyrimidotriazepine derivatives were prepared using the readily obtainable starting material pyrido[2,3-d]pyrimidinthione1 and its hydrazino derivative. The antimicrobial screening of selected synthesized compounds was done using the agar diffusion assay. The IR, 1H NMR and mass spectra of the synthesized compounds were investigated.
Keywords: Fused Pyrimidines, One Carbon Donor, Fused [1,2,4]Triazepine
Cite this paper: Mahmoud R. Mahmoud , Hassan M. F. Madkour , Mohamed M. Habashy , Amr M. El-Shwaf , "Heteroannulation of Pyrido[2,3-d]Pyrimidines. Synthesis and Spectral Characterization of Pyridotriazolopyrimidines, Pyridopyrimidotriazine and Pyridopyrimidotriazepine Derivatives", American Journal of Organic Chemistry, Vol. 2 No. 1, 2012, pp. 39-47. doi: 10.5923/j.ajoc.20120201.08.
1. Introduction
- Pyridopyrimidines were reported to constitute series of compounds which showed broad biological activity such as anticonvulsive, antitumor, antiasthmatic, antiallergic, antihypertensive and useful as diuretic compounds, together with many other applications1-10. As a continuation for our interest for the studies on synthesis of different heterocycles of expected biological activity11-25, the present investigation deals with the synthesis and chemistry of new heteroannulated pyrido[2,3-d]pyrimidine derivatives with the aim of finding new chemotherapeutic agents.
2. Results and Discussions
- The reaction of equimolar portions of m-chloro-α-cyano- cinnamonitrile 1 with 6-aminothiouracil 2 in refluxing ethanol in the presence of a catalytic amount of piperidine afforded 7- amino- 5-(3-chlorophenyl)- 4- oxo- 2-thioxo-1H, 3H-pyrido[2,3-d]pyrimidin-6-carbonitrile 3. The structure 3 was established as pyridopyrimidine rather than thiazinopyrimidine 4 on the basis of IR and 1H-NMR spectra which revealed a pattern completely in accord with the structure 3. (Scheme 1)Thus, the i.r spectrum of 3 exhibited the well defined absorption bands at 3447, 3299, 3216 (νNH2, NH), 2216 (νC≡N), 1696 (νC=O), 1623 (νC=N) and 1170 (νC=S). 1H-NMR spectrum (DMSO-d6) revealed the following signals at δ 12.7 (s, 1H, NH, exchangeable with D2O), 12.2 (s, 1H, NH, exchangeable with D2O), 7.7 (br.s, 2H, NH2, exchangeable with D2O), 7.04-6.8 (m, 4Harom.). The structure 3 was further supported by its mass spectrum which displayed the correct molecular ion peak at m/z = 330 (M, 100%) which is the base peak together with M+1 and M+2 peaks at m/z = 331 (17.6%), 332 (31.2%), respectively.
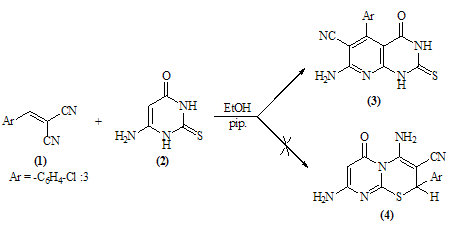 | Scheme 1. |
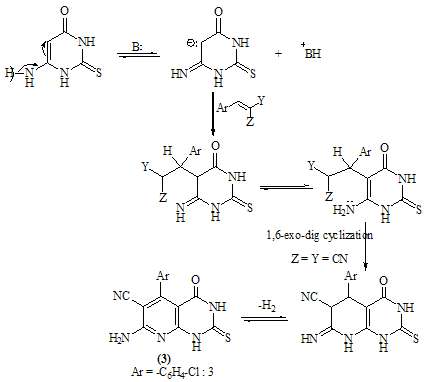 | Scheme 2. |
 | Scheme 3. |
|
|
 | Scheme 4. |
 | Scheme 5. |
 | Scheme 6. |
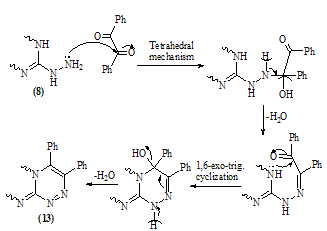 | Scheme 7. |
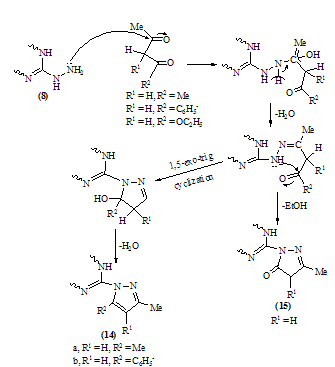 | Scheme 8. |
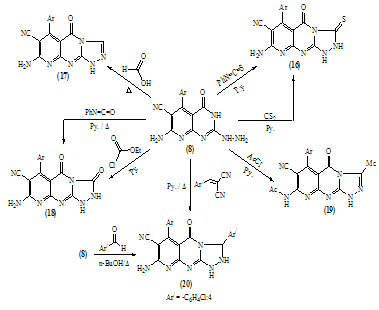 | Scheme 9. Synthesis of pyrido[2,3-d][1,2,4]triazolo[4,3-a]pyrimidine derivatives |
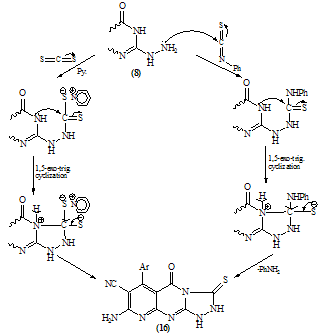 | Scheme 10. |
|
 | Scheme 11. |
4. Experimental
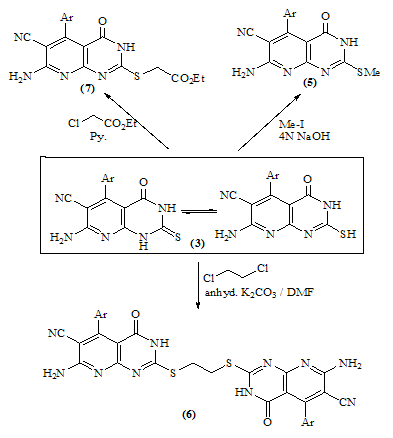
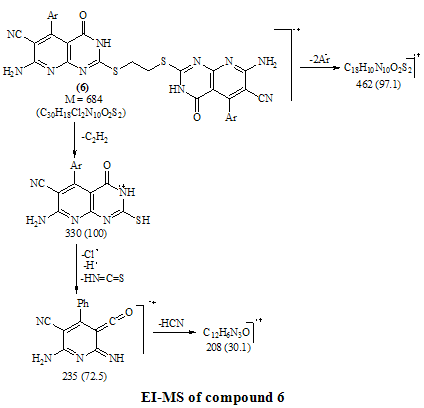
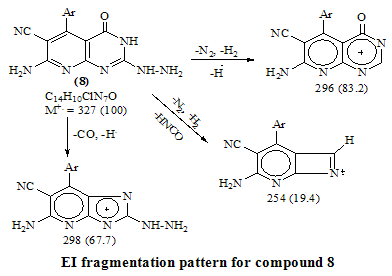
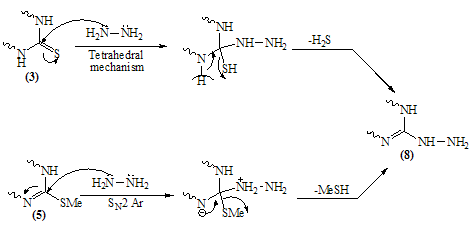
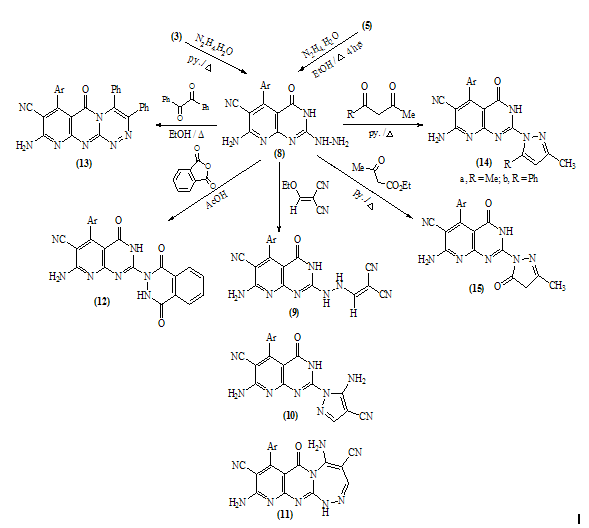
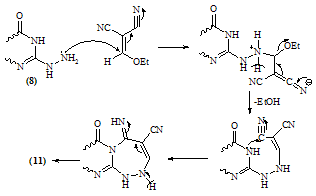
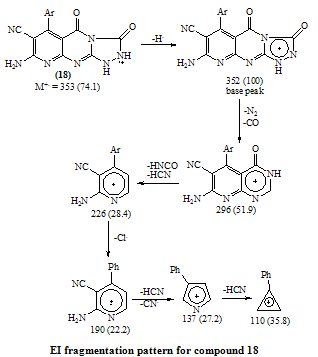
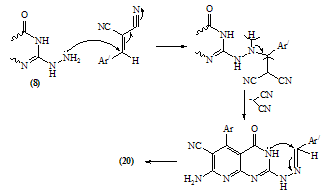
- Melting points are uncorrected and were measured by an electric melting point apparatus (G-K). The IR spectra were recorded on a Pye-Unicam SP1200 spectrophotometer using KBr Wafer technique. The ¹H-NMR spectra were determined on a Varian GEMINI 200 MHz NMR spectrophotometer using CDCl3 or DMSO-d6 as solvent and TMS as an internal standard. All chemical shifts are in ppm downfield from TMS. The elemental analysis were carried out in microanalytical lab of Faculty of Science, . MS were recorded on Shimadzu GC-MS QP1000EX instrument in microanalytical lab, . The monitoring of the progress of all reactions and homogeneity of the synthesized compounds were carried out by TLC.Reaction of 6-aminothiouracil with arylidene malononit- rile: Formation of 7-amino -5-(3–chlorophe-nyl)-4-oxo-2- thioxo-1H,3H-pyrido [2,3-d] pyrimidin- 6-carbonitrile 3A mixture of 6-aminothiouracil (1.43 g, 0.01 mole) and 2-cyano-m-chloro cinnamonitrile (0.01 mole) in ethanol (50 ml) containing piperidine (1 ml) was heated under reflux for 6 hrs (TLC). The deposited solid during reflux was collected by filtration with suction, washed by ethanol, dried and crystallized from dioxane to give 3 as yellow crystals, m.p. 302-4℃, yield 64%. IR (KBr): 3447, 3299, 3216 (νNH2, NH), 2216 (νC≡N), 1696 (νC=O), 1623 (νC=N) and 1170 (νC=S). 1H-NMR (DMSO-d6): δ (ppm) 12.77 (s, 1H, NH, exchangeable with D2O), 12.2 (s, 1H, NH, exchangeable with D2O), 7.7 (br.s, 2H, NH2, exchangeable with D2O), 7.04-6.8 (m, 4Harom.). MS (m/z): 330 (M, 100), 331 (M+1, 17.6), 332 (M+2, 31.2), 305 (28.6), 271 (14.8), 190 (10.5), 84 (83.7), 56 (85.7). Anal. Calcd. for C14H8ClN5OS (329.5): C, 50.98; H, 2.42; N, 21.2; S, 9.7; Cl, 10.77. Found: C, 51.21; H, 2.6; N, 21.06; S, 10.0; Cl, 10.82.Alkylation of 3 using methyl iodide: Formation of 7- amino-5-(3-chlorophenyl)-4-oxo-2-methylthio-3H-pyrido[2,3-d] pyrimidin-6-carbonitrile 5A mixture of 3 (3.3 g, 0.01 mole), methyl iodide (1.4 ml, 0.01 mole) and 4N sodium hydroxide (10 ml) in ethanol (50 ml) was refluxed for 3 hrs. The excess solvent was distilled and the reaction mixture was poured into ice-water and acidified with conc. HCl. The separated solid was filtered off, washed several times with water, dried and recrystallized from ethanol as colourless crystals, m.p. > 300℃, yield 86%. IR (KBr): 3465, 3300, 3230 cm-1 (νNH2), 2214 cm-1 (νC≡N), 1664 cm-1 (νC=O) and 1623 cm-1 (νC=N). 1H-NMR (CDCl3): δ (ppm) 12.12 (s, 1H, NH, exchangeable with D2O), 7.7 (br.s, 2H, NH2, exchangeable with D2O), 7.0-6.8 (m, 4Harom.), 3.3 (s, 3H, S-CH3). MS (m/z): 344 (M+1, 1.7), 345 (M+2, 7.2), 343 (M, 28.4), 302 (17.9), 185 (20.3), 129 (52.4), 97 (96.6), 69 (100). Anal. Calcd. for C15H10ClN5OS (343.5): C, 52.4; H, 2.9; N, 20.37; S, 9.3; Cl, 10.33. Found: C, 52.46; H, 3.12; N, 20.16; S, 9.09; Cl, 10.11.Reaction of 3 with 1,2-dichloroethane: Formation of Bis- S-[7-amino-5-(3-chloro-phenyl)-6-cyano-3,4-dihydro–pyrido[2,3-d]pyrimidin-2-yl]ethan-1,2-dithiol 6To a solution of 3 (0.01 mole) in DMF (50ml), 1,2-dichloroethane (0.01 mole) was added with stirring in the presence of anhydrous K2CO3 (2.5 g). The reaction mixture was heated under reflux for 12 hrs (TLC), then poured into water and stirred for 30 min. The deposited was filtered off, dried then crystallized from DMF to give 6 as yellow crystals, m.p. > 300℃, yield 34%. IR (KBr): 3452, 3343, 3208 (br.) cm-1 (νNH2), 2213, 2176 cm-1 (νC≡N), 1696, 1672 cm-1 (νC=O) and 1623 cm-1 (νC=N). 1H-NMR (DMSO-d6): δ (ppm) 12.4 (br.s, 1H, NH, exchangeable with D2O), 11.8 (s, 1H, NH, exchangeable with D2O), 7.77-7.3 (m, 8Harom.), 4.2 (br.s, 4H, SCH2CH2S), 2.6 (br.s, 4H, exchangeable with D2O). MS (m/z): 462 [(M-2Ar.), 97.1], 331 (100), 235 (72.5), 208 (30.1). Anal. Calcd. for C30H18Cl2N10O2S2 (684): C, 52.63; H, 2.63; N, 20.46; S, 9.35. Found: C, 52.34; H, 2.9; N, 20.09; S, 9.0.Alkylation of 3 using ethyl chloroacetate: Formation of 7-amino– 5- (3-chlorophenyl)- 2- ethoxycarbony lmethy- lthio- 4- oxo-3,4- dihydro- pyrido[2,3-d]pyrimidin- 6- carbonitrile 7A mixture of compound 3 (0.01 mole), ethyl chloroacetate (0.01 mole) in pyridine (30 ml) was refluxed for 6 hrs (TLC). The reaction mixture was poured into water and then acidified with ice cold HCl. The separated colourless product was collected by filtration, dried and then recrystallized from DMF to give 7 as colourless crystals, m.p. 304-6℃, yield 52.6%. IR (KBr): 3470(w), 3300(w), 3170 cm-1 (νNH2), 2208 cm-1 (νC≡N) and 1667 cm-1 (νC=O). 1H-NMR (DMSO-d6): δ (ppm) 12.5 (s, 1H, NH, exchangeable with D2O), 7.63 (br.s, 2H, NH2, exchangeable with D2O), 7.03-6.8 (m, 4Harom.), 4.2 (q, 2H, J = 7.2 Hz), 3.8 (s, 2H, SCH2COOEt), 1.2 (t, 3H, J = 7.2 Hz). MS (m/z): 380 [M-Cl., 20.8]. Anal. Calcd. for C18H14ClN5O3S (415.5): C, 51.98; H, 3.36; N, 16.84; S, 7.7. Found: C, 52.12; H, 3.16; N, 16.7; S, 7.53.Reaction of 3 and 5 with hydrazine hydrate: Formation of 7-amino-5-(3-chloro-phenyl)-2-hydrazino-4-oxo-3, 4- dihydropyrido[2,3-d]pyrimidin-6-carbonitrile 8A mixture of compound 3 or 5 (0.01 mole) and hydrazine hydrate 80% (0.5 ml, 0.01 mole) in absolute ethanol (50 ml) was heated under reflux for 6 hrs (TLC). The white product that deposited during reflux was collected by suction and washed by ethanol then recrystallized from dioxane to give 8 as colourless crystals, m.p. 332-4℃, yield 22.6% and 58.2%, respectively. IR (KBr): 3460, 3335, 3218 cm-1 (νNH2, NH), 2208 cm-1 (νC≡N), 1692 cm-1 (νC=O) and 1652 cm-1 (νC=N). 1H-NMR (DMSO-d6): δ (ppm) 9.66 (s, 1H, NH, exchangeable with D2O), 8.9 (m, 4Harom.), 5.2-4.8 (Humb, 5H, exchangeable with D2O). MS (m/z): 327 (M, 100), 328 (M+1, 12.6), 329 (M+2, 54.5), 298 (67.7), 296 (83.2), 254 (19.4). Anal. Calcd. for C14H10ClN7O (327.5): C, 51.29; H, 3.05; N, 29.92. Found: C, 51.54; H, 2.91; N, 29.76.Reaction of 8 with ethoxymethylene malononitrile: Formation of 5,10-diamino-4,9-dicyano-8- (3-chloroph - enyl) - 1H-pyrido [2,3-d]pyrimido[2,1-c] 1,2,4-triazepine -7(H)-one 11A mixture of 8 (0.01 mole) and ethoxymethylene malono- nitrile (0.01 mole) in pyridine (30 ml) was heated under reflux for 6 hrs. The solid separated on hot was filtered off, washed several times with ethanol, dried and recrystallized from ethanol as pale yellow crystals, m.p. 266-8℃, yield 57.3%. IR (KBr): 3406, 3323, 3222, 3172 cm-1 (νNH2, NH), 2225 cm-1 (νC≡N), 1685 cm-1 (νC=O) and 1641 cm-1 (νC=N). MS (m/z): 403 (M, 62.7), 404 (M+1, 39.2), 405 (M+2, 20.7), 402 (M-1, 100). Anal. Calcd. for C18H10ClN9O (403.5): C, 53.53; H, 2.47; N, 31.22. Found: C, 53.62; H, 2.73; N, 31.50.Synthesis of N-[7-amino-5-(3-chlorophenyl)- 6-cyano-4 -oxo-3H -pyrido[2,3-d]pyrimid- in -2- yl]phthalizindione 12A mixture of compound 8 (0.006 mole) and phthalic anhydride (0.006 mole) in acetic acid (30 ml) was heated under reflux for 3 hrs (TLC). The excess acetic was collected by distillation and left a semi solid product which triturated with ethanol and the precipitate was filtered off, dried and recrystallized from methanol to give the phthalizindione derivative 12 as pale yellow crystals, m.p 280-2℃, yield 69.3%. IR (KBr): 3388 (br.), 3144 cm-1 (νNH2,NH), 2215 cm-1 (νC≡N), 1715 cm-1 (νC=O), 1630 cm-1 (νC=N). MS (m/z): 458 (M, 35.1), 456 (M-H2, 100). Anal. Calcd. for C22H12ClN7O3 (457.5): C, 57.7; H, 2.62; N, 21.42. Found: C, 57.99; H, 2.48; N, 21.35.Reaction of 8 with 1,2-diketone: Formation of 9-amino-7-(3-chlorophenyl)-3,4-diphenyl -6-oxo - pyrido [2 ,3 -d] pyrimido [2,1-c]- 1,2,4-triazin-8-carbonitrile 13Benzil (0.01 mole) was added to a solution of compound 8 (0.01 mole) in dioxane (30 ml) and the reaction mixture was heated under reflux for 8 hrs. The solid separated on hot was filtered off, dried and recrystallized from DMF to give the pyridopyrimidotriazine derivative 13 as brown crystals, m.p 350-2℃, yield 28.6%. IR (KBr): 3460, 3334, 3217 cm-1 (νNH2,NH), 2209 cm-1 (νC≡N), 1688 cm-1 (νC=O), 1656 cm-1 (νC=N). MS (m/z): 347 (M-2Ph., 12.4), 327 (M-Ph-C≡C-Ph, 100). Anal. Calcd. for C28H16ClN7O (501.5): C, 66.99; H, 3.19; N, 19.54. Found: C, 67.04; H, 3.21; N, 19.38.Reaction of 8 with 1,3-diketones: Formation of 7- amino -5-(3- chlorophenyl)- 2-(3,5- dimethylpyrazol- 1-yl) - 3,4- dihydropyrido[2,3-d] pyrimidin-6-carbonitrile 14a and 7-amino-5-(3-chlorophenyl)- 2-(3- methyl-5- phenyl– pyrazol-1-yl)4-oxo-3,4-dihydropyrido-[2,3-d]pyrimidin- 6-carbonitrile 14bTo a solution of compound 8 (3.3 g, 0.01 mole) in pyridine (30 ml), pentan-2,4-dione and/or 1-phenyl-butan-1,3-dione (0.01 mole) was added and the reaction mixture was refluxed for 6 hrs (TLC). After cooling, the content was diluted with water and acidified with ice cold acetic acid. The precipitated solid was collected by suction, dried and recrystallized from the proper solvent to give 14a and 14b, respectively.7-amino-5-(3-chlorophenyl)-2-(3,5-dimethylpyrazol-1-yl)-3,4-dihydropyrido[2,3-d]pyrimidin-6-carbonitrile 14a7-amino-5-(3-chlorophenyl)-2-(3-methyl-5-phenylpyrazol-1-yl)-4-oxo-3,4-dihydropyrido[2,3-d]pyrimidin-6-carbonitrile 14bRecrystallized from dilute methanol as yellowish-white crystals, m.p. 287-9℃, yield 69%. IR (KBr): br. centered at 3153 cm-1 (νNH2, NH), 2218 cm-1 (νC≡N), 1678 cm-1 (νC=O) and 1632 cm-1 (νC=N). 1H-NMR (DMSO-d6): δ (ppm) 12.03 (s, 1H, NH, exchangeable with D2O), 8.09-8.06 (d, 1H, J = 8.7 Hz), 7.74 (br.s, 2H, NH2, exchangeable with D2O), 7.4-6.9 (m, 9Harom.), 2.79 (s, 3H, C3’-Me). MS (m/z): 343 [M-Ar., 100]. Anal. Calcd. for C24H16ClN7O (453.5): C, 63.5; H, 3.52; N, 21.6. Found: C, 63.76; H, 3.61; N, 21.8.Reaction of 8 with β-ketoester: Formation of 7–amino-5- (3-chlorophenyl)-2-(3-methyl-pyrazol-5-on-1-yl)-4-oxo- 3,4 -dihydropyrido[2,3-d] pyrimidin-6- carbonitrile 15A mixture of hydrazinopyridopyrimidine 8 (0.01 mole) and ethyl acetoacetate (0.01 mole) in pyridine (30 ml) was refluxed for 8 hrs (TLC). The excess solvent was collected by distillation and the whole mixture was acidified with ice cold acetic acid. The solid deposited was filtered off, washed several times with water, dried and recrystallized from dioxane to give 15 as pale yellow crystals, m.p. 273-6℃, yield 57%. IR (KBr): 3324, 3207, 3158 cm-1 (νNH2, NH), 2210 cm-1 (νC≡N), 1668 cm-1 (νC=O). MS (m/z): 380 (M-Me.), 344 (M-Me., Cl., 100). Anal. Calcd. for C18H12ClN7O2 (393.5): C, 54.89; H, 3.04; N, 24.9. Found: C, 54.92; H, 2.95; N, 24.46.Reaction of compound 8 with phenyl isothiocyanate and/or carbon disulphide: Formation of 8-amino-6- (3- chlorophenyl)-7-cyano-5-oxo-1H-1,2,4-triazolo[4,3-a] pyrido[2,3-d]pyrimidin-3(H)thione 16Method 1:A mixture of compound 8 (0.01 mole) and phenyl isothiocyanate (0.01 mole) in pyridine (20 ml) was stirred with reflux for 2 hrs (TLC). The reaction mixture poured onto water and acidified with dilute acetic acid. The dark crude product was collected and washed several times with dilute ethanol, dried and recrystallized from dioxane to give 16 as light brown crystals, m.p 340-2℃, yield 47.2%. IR (KBr): 3391, 3327, 3230 cm-1 (νNH2, NH), 2222 cm-1 (νC≡N), 1726, 1654 cm-1 (νC=O), 1234 cm-1 (νC=S). MS (m/z): 369 (M, 100), 337 (82.3), 296 (25.4), 286 (22), 224 (18.7). Anal. Calcd. for C15H8ClN7OS (369.5): C, 48.71; H, 2.16; N, 26.52; S, 8.66. Found: C, 48.69; H, 2.19; N, 26.31; S, 8.55.Method 2:To a solution of compound 8 (2 g, 0.006 mole) in pyridine (15 ml), carbon disulphide (5 ml) was added with stirring for 10 min., then the reaction mixture was refluxed for 18 hrs on water bath until all the hydrogen sulphide gas was evolved. The mixture was acidified with ice cold acetic acid. The precipitate was filtered off, dried and recrystallized from dioxane to give 16, yield 56.3% (identity m.p, mixed m.p, TLC and IR comparison).Reaction of 8 with formic acid: Formation of pyrido- triazolopyrimidine derivative 17Compound 8 (2 g, 0.006 mole) was heated under reflux with formic acid (10 ml) for 6 hrs (TLC). After cooling, the reaction mixture was poured into distilled H2O (50 ml). The precipitated was collected by filtration, dried and then recrystallized from ethanol to give 17 as pale yellow crystals, m.p. 316-8℃, yield 62%. IR (KBr): br. 3312, br. 3165 cm-1 (νNH2), 2227 cm-1 (νC≡N), 1706 cm-1 (νC=O) and 1634 cm-1 (νC=N). MS (m/z): 338 (M, 57.3), 337 (M-1, 100), 339 (M+1, 45.8), 340 (M+2, 19.1), 310 (26.7), 225 (16.8), 199 (17.9), 163 (10.7). Anal. Calcd. for C15H8ClN7O (337.5): C, 53.33; H, 2.37; N, 29.03. Found: C, 53.42; H, 2.44; N, 28.83.Reaction of 8 with ethyl chloroformate and/or phenyl isocyanate: Formation of pyridotriazolopyrimidine 18Method 1:A mixture of hydrazinopyridopyrimidine 8 (0.01 mole) and ethyl chloroformate (0.01 mole) in pyridine (20 ml) was refluxed for 8 hrs. The solid separated on hot was filtered off, washed several times with water, dried and recrystallized from ethanol to give 18 as light brown crystals, m.p. 246-50℃, yield 79%. IR (KBr): 3334, 3221 cm-1 (br.) (νNH2, NH), 2216 cm-1 (νC≡N), 1706 (w), 1685 cm-1 (νC=O) and 1618 cm-1 (νC=N). MS (m/z): 353 (M, 74.1), 354 (M+1, 44.4), 355 (24.7), 352 (M-1, 100), 296 (51.9), 226 (28.4), 190 (22.2), 137 (27.2), 110 (35.8). Anal. Calcd. for C15H8ClN7O2 (353.5): C, 50.92; H, 2.26; N, 27.72. Found: C, 50.76; H, 2.11; N, 27.56. Method 2:To a solution of compound 8 (0.01 mole) in pyridine (20 ml), phenyl isocyanate (0.01 mole) was added and the reaction mixture was heated under reflux for 6 hrs. The separated solid on hot collected by filtration, washed with water, dried and recrystallized from ethanol to give 18 (identity m.p, mixed m.p, TLC and IR comparison). The mother liquor when diluted with H2O gave a colourless solid which collected by filtration and identified as sym. diphenyl urea [M = 212 (10.9)].Reaction of 8 with acetyl chloride: Formation of 8-acetylamino- 6-(3-chlorophenyl)- 3-methyl-5-oxo- 1H- pyrido [2,3- d] 1,2,4-triazolo[4,3-a] pyrimidin-7-carbo- nitrile 19To a solution of compound 8 (2 g, 0.006 mole) in pyridine (15 ml), acetyl chloride (10 ml) was added slowly in ice bath with stirring for 5 min. then the reaction mixture was refluxed on water-bath for 30 min. Pouring the reaction content on water left a crude solid product which filtered off, washed with dilute methanol and recrystallized from dioxane to give 19 as bage crystals, m.p 218-220℃, yield 70%. IR (KBr): 3326, 3128 cm-1 (νNH), 2218 cm-1 (νC≡N), 1720, 1704 cm-1 (νC=O). 1H-NMR (CDCl3): δ (ppm) 11.06 (s, 1H, NH, exchangeable with D2O), 7.6-7.3 (m, 4Harom.), 3.3 (br.s, 1H, exchangeable with D2O), 2.4 (s, 3H, C3-Me), 2.1 (s, 3H, COMe). MS (m/z): 393 (M, 8.3), 351 (M-CH2=C=O, 100), 310 (22.4), 255 (12.6), 199 (14.1). Anal. Calcd. for C18H12ClN7O2 (393.5): C, 54.89; H, 3.04; N, 24.9. Found: C, 55.07; H, 2.99; N, 24.67.8-Amino-3-(4-chlorophenyl)-6-(3-chlorophenyl)-5-oxo-1H,2H,3H-pyrido - [2,3 - d] 1 , 2,4 - triazolopyrimidin–7- carbonit - rile 20A mixture of compound 8 (0.01 mole) and α-cyano- 4-chlorocinnamonitrile (0.01 mole) in pyridine (20 ml) was heated under reflux for 7 hrs. The solid separated on hot was filtered off, washed several times with ethanol, dried and then recrystallized from benzene as bage crystals, m.p. 236-8℃, yield 82%. IR (KBr): 3354, 3187 cm-1 (νNH2, NH), 2216 cm-1 (νC≡N), 1716 cm-1 (w) (νC=O) and 1647 cm-1 (νC=N). MS (m/z): 450 (M, 28.3), 451 (M+1, 7.6), 452 (M+2, 6.3), 338 (M-Ar’, 100), 180 (10.9), 124 (17.6), 89 (27.9). Anal. Calcd. for C21H13Cl2N7O (450): C, 56.0; H, 2.88; N, 21.77; Cl, 15.77. Found: C, 56.13; H, 3.1; N, 21.52; Cl, 15.44.Authentic sample of 20A mixture of compound 8 (0.9 g, 0.002 mole) and 4- chlorobenzaldehyde (0.002 mole) was dissolved in n-butanol (20 ml) and refluxed for 2 hrs. The excess n-butanol was removed by distillation and left a yellowish-white solid which recrystallized from benzene to give 20 (identity m.p., mixed m.p., TLC and IR comparison).