-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2012; 2(1): 35-38
doi:10.5923/j.ajoc.20120201.07
Efficient Route to (2R)-6-Hydroxy-2-methyl- dihydropyridin-3-ones, Key Intermediates for Piperidine Alkaloids Syntheses
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLEvangelia N. Tzanetou1, Konstantinos M. Kasiotis2, Serkos A. Haroutounian1
1Chemistry Laboratory, Agricultural University of Athens, Iera Odos 75, Athens, 11855, Greece
2Benaki Phytopathological Institute, Laboratory of Pesticides Toxicology, 8 St. Delta Street, Athens, Kifissia, 14561, Greece
Correspondence to: Serkos A. Haroutounian, Chemistry Laboratory, Agricultural University of Athens, Iera Odos 75, Athens, 11855, Greece.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
The efficient synthesis of (2R,6S)-6-hydroxy-2-methyl-1-tosyl-1,2-dihydropyridin-3(6H)-one 5 in five steps and 30 % overall yield is reported. This dihydropyridone constitutes versatile chiral building block repetitively used in the synthesis of various piperidine alkaloids and may serve as key template for the construction of synthetic libraries of bioactive derivatives and analogues of the piperidine alkaloids.
Keywords: Pyridones, Piperidine Alkaloids, Azasugars, Glucal
Cite this paper: Evangelia N. Tzanetou, Konstantinos M. Kasiotis, Serkos A. Haroutounian, Efficient Route to (2R)-6-Hydroxy-2-methyl- dihydropyridin-3-ones, Key Intermediates for Piperidine Alkaloids Syntheses, American Journal of Organic Chemistry, Vol. 2 No. 1, 2012, pp. 35-38. doi: 10.5923/j.ajoc.20120201.07.
1. Introduction
- Multifunctionalized piperidine alkaloids (including azasugars) constitute a distinct class of natural compounds widely found in living systems[1] that exhibit a wide range of biological activities, as a result of their ability to mimic carbohydrate substrates in a plethora of enzymatic processes [2,3]. These activities include glycosidase inhibition[4], tumour growth inhibition[5] and anti-HIV behaviour[6], making those particularly attractive synthetic targets. Thus, an intense research effort has been devoted for the development of methodologies and synthetic strategies for the efficient approach of these compounds and their derivatives[7-10]. Among the various synthetic routes developed for their efficient access, the use of dihydropyridones as advanced intermediates −prior to their conversion to piperidines− constitutes a well reviewed important synthetic strategy[11,12] that has been a long term objective of our research group [12 and references therein]. In this respect, a series of synthetic methods including the microwave- assisted synthesis, regioselective nucleophilic addition to pyridinium salts, incorporation of boronate esters to dihydropyridones, have been reported during the last 5 years as concise routes for the preparation of dihydropyridone derivatives[13-15].Herein we report the efficient transformation of the commercially available 3,4-di-O-acetyl-6-deoxy-L-glucal 1 to (2R, 6S)-2-methyl-dihydropyridone 5, which accounts as the key intermediate for the synthesis of many bioactive natural piperidine alkaloids, such as the molecules of (−)-Cassine, (−)-Spectaline, (−)-Carnavaline, Prosafrine and (−)-Prosafrinine. The literature reports implicating the derivatives of L-glucal in synthetic endeavours are limited and mainly refer to their transformation to L-ristosamine and L-epi-daunosamine glycosides[16].
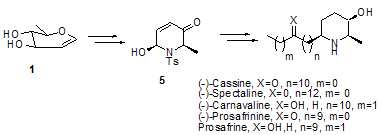 | Figure 1. Piperidine alkaloids approached through the synthesis of key intermediate dihydropyridone 5 |
2. Chemistry
- Our synthetic route substrate is the commercially available molecule of 3,4-di-O-acetyl-6-deoxy-L-glucal, which in acidic environment (0.002 M H2SO4 solution) by reaction with HgSO4was efficiently converted to the optically active furfural 2, in accordance to a previously reported method [17]. The displacement of the secondary hydroxy group with the azide moiety, with the simultaneous inversion of its configuration, was performed in high enantiomeric excess (>98%) by treating the compound 2 with DBU in toluene and DPPA. The enantiomeric excess of azide 3 was determined after the hydrogenation of the molecule for 50 min over 10% Pd/C catalyst and 1 bar pressure[18]. The mixture was filtered over Celite® and the resulting amine was in situ derivatized with (−)-menthyl chloroformate, in the presence of Et3N. The enantiomeric ratio was determined by reversed- phase HPLC [Kromasil 100-5, C-18, H2O/MeOH/ CH3CN gradient elution from 45:35:20 to 0:20:80, flow = 1.1 mL/min, UV detection at 238 nm]; tR major 49.2 min (97%); and tR minor 50.1 min (3%).
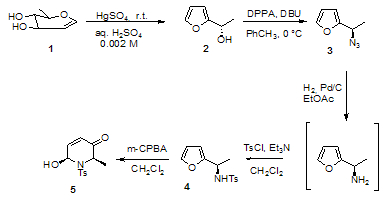 | Scheme 1. Reagents and Conditions (a) DPPA, DBU, toluene, 0 °C; (b) H2, Pd/C, EtOAc; (c) TsCl, Et3N, CH2Cl2; (d) m-CPBA, CH2Cl2 |
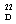 = –24.4) constitutes an additional proof of the assigned configuration, since its enantiomer displays the opposite sign ([α]
= –24.4) constitutes an additional proof of the assigned configuration, since its enantiomer displays the opposite sign ([α] 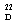 = +24.9).
= +24.9).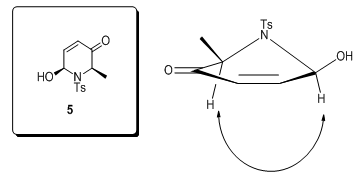 | Figure 2. NOE correlation in dihydropyridone 5 |
3. Experimental Part
- Air- and /or moisture sensitive reactions were carried out under argon atmosphere in flame-dried glassware. Solvents were distilled from the appropriate drying agents prior to use. All starting materials were purchased from Aldrich (analytical reagent grades) and used without further purification.All reactions were monitored by thin-layer chromatogra-phy using TLC sheets coated with silica gel 60 F254 (Merck); spots were visualized with UV light or by treatment with. Products were purified by flash chromatography on Merck silica gel 60 (230-400 mesh ASTM). Melting points (un-corrected): Büchi melting point apparatus. FT-IR: Nicolet Magna 750, series II. Samples were recorded as KBr pellets, unless otherwise stated. 1H NMR spectra were recorded on a Bruker DRX-400 (400 MHz) spectrometer, in the indicated solvents. Chemical shifts are referenced to internal TMS.HPLC measurements were performed using an Agilent 1100 series instrument equipped with a variable UV wave-length detector and coupled to HP ChemStation utilizing the manufacturer’s 5.01 software package.
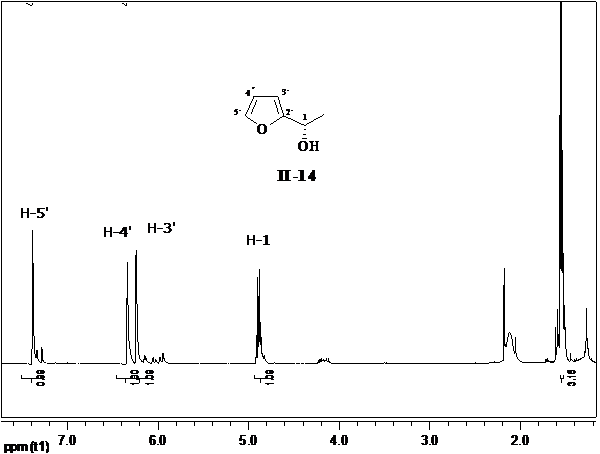 | Figure 3. 1H NMR of compound 2 |
4. Conclusions
- The commercially available L-glucal substrate provided efficiently the molecule of dihydropyridin-3(6H)-one in five steps and satisfactory overall yield (30.3%). This compound can serve as crucial key intermediate for the efficient syntheses of various bioactive natural piperidine alkaloids.