-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2011; 1(1): 14-20
doi: 10.5923/j.ajoc.20110101.04
Synthesis and Spectral Characterization of Novel Pyridotriazolo-, Pyridothiazolo-, Pyridotetrazolopyrimidines and Pyridopyrimidotriazepine Derivatives for Potential Pharmacological Activities
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLMahmoud R. Mahmoud 1, Manal M. El-Shahawi 1, Fatma S.M. Abu El-Azm 1, Samira E. Farahat 2
1Chemistry Department, Faculty of Science, Ain Shams University, Abbassia 11566, Cairo, Egypt
2Chemistry Department, Faculty of Science, 7th of April University, Gharian, Libye
Correspondence to: Fatma S.M. Abu El-Azm , Chemistry Department, Faculty of Science, Ain Shams University, Abbassia 11566, Cairo, Egypt.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Number of pyridotriazolo-, pyridothiazolo-, pyridotetrazolopyrimidines and pyrido-pyrimidotriazepine derivatives were prepared using the readily obtainable starting material pyrido[2,3-d]pyrimidinthione1 and its hydrazino derivative. The antimicrobial screening of selected synthesized compounds was done using the agar diffusion assay. The IR, 1H NMR and mass spectra of the synthesized compounds were investigated.
Keywords: Fused Pyrimidines, One Carbon Donor, Fused [1,2,4]Triazepine
Cite this paper: Mahmoud R. Mahmoud , Manal M. El-Shahawi , Fatma S.M. Abu El-Azm , Samira E. Farahat , "Synthesis and Spectral Characterization of Novel Pyridotriazolo-, Pyridothiazolo-, Pyridotetrazolopyrimidines and Pyridopyrimidotriazepine Derivatives for Potential Pharmacological Activities", American Journal of Organic Chemistry, Vol. 1 No. 1, 2011, pp. 14-20. doi: 10.5923/j.ajoc.20110101.04.
Article Outline
1. Introduction
- [1,2,4]Triazoles represent a class of heterocyclic compounds of significant importance in a agriculture and medicine2,3. They used in metalloorganic chemistry as polyfunctional lignads4. They also exhibit a broad spectrum of biological activity.5 Pronounced pharmacological and biological activities are also intrinsic for pyridopyrimidines6-10. This stimulated us to combine the above pharmacophoric fragments is a single molecule and to continue our previous work for synthesis of novel heterocycles.11-21 Here, we used the easy obtainable substituted chalcone and 6-aminothiouracil for synthesis of 5-(4-methylphenyl)-7-phenyl-2-thioxo-2,3- dihydropyrido[2,3-d]pyrimidin-4(3H)one1 1.
2. Results and Discussions
- 6-Aminothiouracil reacted with 3-(4-methoxyphenyl)- 1-phenylprop-2-enone in equimolar portion in the presence of catalytic amount of piperidine in ethanol to afford the adduct 2,3-dihydro-5-(4-methoxyphenyl)-7-phenyl-2-thioxo pyrido[2,3-d]pyrimidin-4(1H)-one 1. The formation of 1 is assumed to proceed via Michael addition of aminothiouracil C-5 to the activated double bond in prop-2-enone derivative, followed by1,6-exo-trig cyclization. 1H NMR spectrum of 1 [DMSO-d6] revealed the signals at δ (ppm): 13.0 (s, 1H, NH, exchangeable with D2O), 12.3 (s, 1H, NH, exchangeable with D2O), 8.4 (s, 1H, C6-H), 8.29-6.9 (m, 9Harom.) and 3.7 (s, 3H, OMe). Moreover, the mass spectrum show fragmentation pattern which completely in accord with the proposed structure 1.
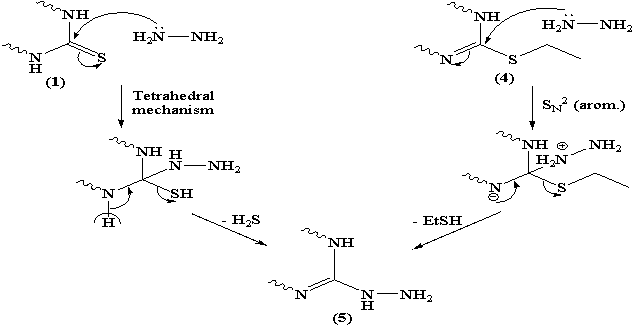
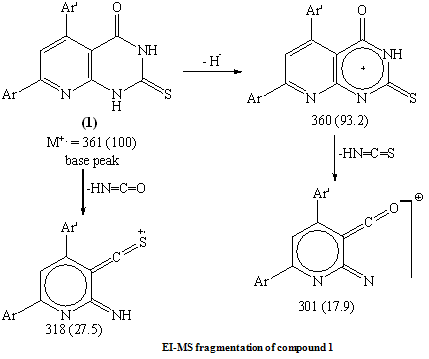 The reaction of pyrido[2,3-d]pyrimidine-2-thione derivative with ethyl chloroacetate was claimed to afford the thioacetic ester derivative 2.22 Here in, refluxing 1 with ethyl chloroacetate in n-butanol in presence of anhydrous sodium acetate yielded 2-carboxymethylthio-5-(4-methoxyphenyl) -7-phenyl-3H,4H-pyrido[2,3-d]pyrimidin-4(3H)one 2. (Scheme 1) The structure features of 2 was elucidated according to the following arguments: i- The IR spectrum devoid νCO (ester) but retained νCO (acid) at 1698 cm-1 together with broad absorption band for –OH group centered at 3446 cm-1. ii- The highest recorded peak in the mass spectrum at m/z = 419 (100) which attributable for the molecular ion and the base peak. (C.f Exp.) iii- Compound 2 easily soluble in sodium bicarbonate solution. iv- Furthermore, 1H NMR spectrum show down field signal attributable for carboxylic acid group at δ 11.3 ppm together with singlet at δ 3.9 ppm integrated for 2H (CH2COOH).Independent chemical proof for compound 2 seemed necessary, thus, when 2 was refluxed with freshly distilled acetic anhydride afforded pyrido [2,3-d] thiazolo [3,2-a] pyrimidine derivative 3. Compound 3 was obtained in fairly good yield by stirring compound 1 with chloroacetyl chloride in pyridine for one hour. Ethylation of 1 using ethyl iodide in ethanol in the presence of fused sodium acetate yielded the S-alkylated product 4. The microanalysis and mass spectrum of 4 indicates incorporation of one mole of ethyl iodide in the reaction product. EI-MS fragmentation show the correct molecular ion peak at m/z = 389 (18.1%) together with the base peak at m/z = 361 (100%) corresponding to the radical cation [M-C2H4]. Moreover, the 1H NMR spectrum show the signals attributable for the S-ethyl group at δ 3.15 ppm as quartet integrated for 2H and δ 1.36 ppm as triplet integrated for 3H. Hydrazinloysis of pyridopyrimidin-2-thione 1 with hydrazine hydrate (80%) in boiling ethanol afforded the sulfur free compound 5 (yield=39.6%). Compound 5 which identified as 2-hydrazino-5-(4-methoxyphenyl)-7-phenyl-4-oxo-3H,4H-pyrido [2,3-d] pyrimidine could be obtained in fairly good yield (56.6%) via nucleophilic displacement of thioethyl group with hydrazine in boiling ethanol. (Scheme 1)The formation of 5 could be visualized as shown in scheme 2.The structure 5 deduced from the satisfactory IR, 1H NMR and MS spectra. The highest recorded m/z value at m/z = 359 (100%) attributable for the molecular ion peak which is the base peak.
The reaction of pyrido[2,3-d]pyrimidine-2-thione derivative with ethyl chloroacetate was claimed to afford the thioacetic ester derivative 2.22 Here in, refluxing 1 with ethyl chloroacetate in n-butanol in presence of anhydrous sodium acetate yielded 2-carboxymethylthio-5-(4-methoxyphenyl) -7-phenyl-3H,4H-pyrido[2,3-d]pyrimidin-4(3H)one 2. (Scheme 1) The structure features of 2 was elucidated according to the following arguments: i- The IR spectrum devoid νCO (ester) but retained νCO (acid) at 1698 cm-1 together with broad absorption band for –OH group centered at 3446 cm-1. ii- The highest recorded peak in the mass spectrum at m/z = 419 (100) which attributable for the molecular ion and the base peak. (C.f Exp.) iii- Compound 2 easily soluble in sodium bicarbonate solution. iv- Furthermore, 1H NMR spectrum show down field signal attributable for carboxylic acid group at δ 11.3 ppm together with singlet at δ 3.9 ppm integrated for 2H (CH2COOH).Independent chemical proof for compound 2 seemed necessary, thus, when 2 was refluxed with freshly distilled acetic anhydride afforded pyrido [2,3-d] thiazolo [3,2-a] pyrimidine derivative 3. Compound 3 was obtained in fairly good yield by stirring compound 1 with chloroacetyl chloride in pyridine for one hour. Ethylation of 1 using ethyl iodide in ethanol in the presence of fused sodium acetate yielded the S-alkylated product 4. The microanalysis and mass spectrum of 4 indicates incorporation of one mole of ethyl iodide in the reaction product. EI-MS fragmentation show the correct molecular ion peak at m/z = 389 (18.1%) together with the base peak at m/z = 361 (100%) corresponding to the radical cation [M-C2H4]. Moreover, the 1H NMR spectrum show the signals attributable for the S-ethyl group at δ 3.15 ppm as quartet integrated for 2H and δ 1.36 ppm as triplet integrated for 3H. Hydrazinloysis of pyridopyrimidin-2-thione 1 with hydrazine hydrate (80%) in boiling ethanol afforded the sulfur free compound 5 (yield=39.6%). Compound 5 which identified as 2-hydrazino-5-(4-methoxyphenyl)-7-phenyl-4-oxo-3H,4H-pyrido [2,3-d] pyrimidine could be obtained in fairly good yield (56.6%) via nucleophilic displacement of thioethyl group with hydrazine in boiling ethanol. (Scheme 1)The formation of 5 could be visualized as shown in scheme 2.The structure 5 deduced from the satisfactory IR, 1H NMR and MS spectra. The highest recorded m/z value at m/z = 359 (100%) attributable for the molecular ion peak which is the base peak.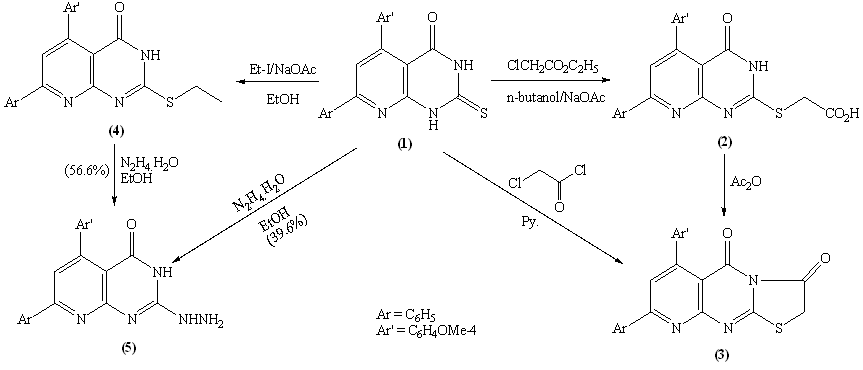 | Scheme 1. |
 | Scheme 2. |
 | Scheme 3. a, CH(OEt)3/Ac2O; b, ClCO2Et/AcOH; c, PhNCS/py.; d, Ac2O |
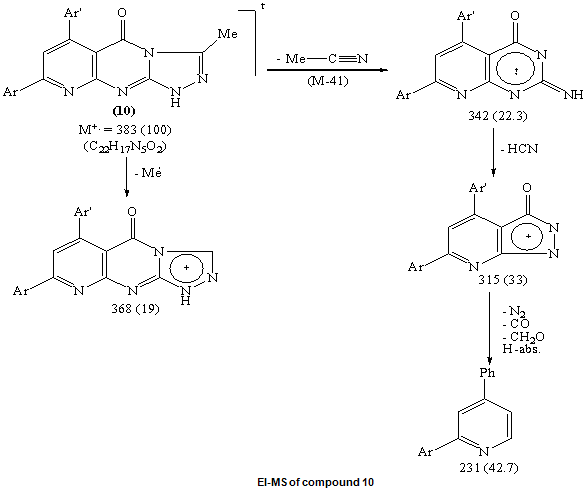
 | Scheme 4. Synthesis of pyridotriazolopyrimidines 8, 9, 10 |
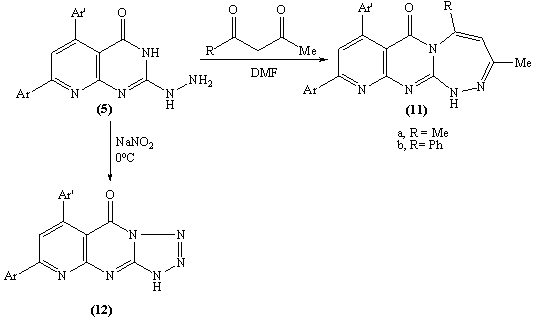 | Scheme 5. |
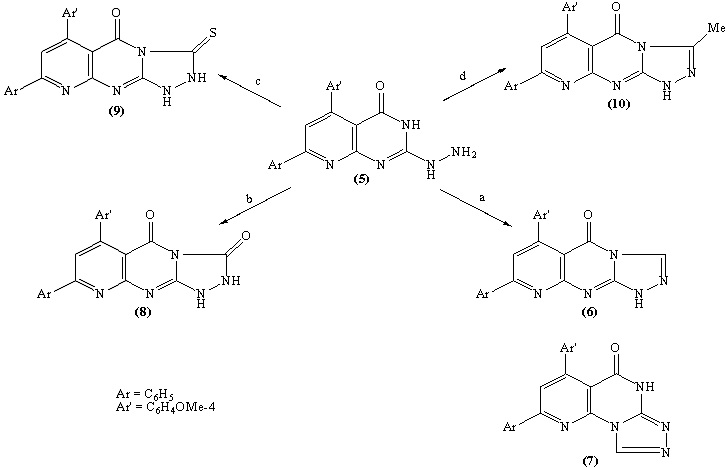
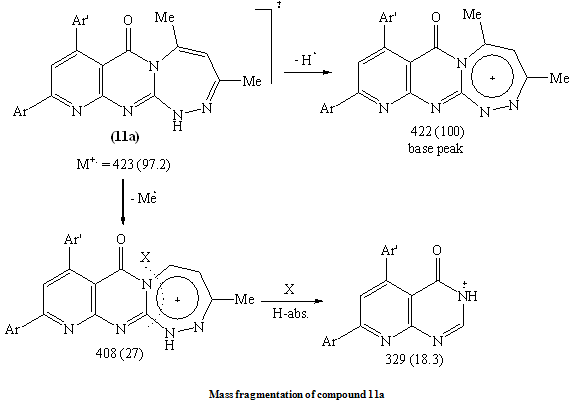 Ample evidence for the structure 9 is forthcoming from 1H NMR analysis which completely consistent with the proposed structure (C.f. Exp.). Treatment of compound 5 with freshly distilled acetic anhydride yielded 3-methyl-8-phenyl -6-(4-methoxyphenyl)-1H,2H-pyrido[2,3-d]1,2,4-triazolol[4,3-a]pyrimidin-5-one 10 whose structure was deduced from the correct analytical and spectroscopic data. The IR spectrum of 10 displayed one absorption band for the carbonyl group at 1699 cm-1 and 1H NMR spectrum [DMSO-d6] revealed signals at δ (ppm) 8.6 (s, 1H, C7-H), 8.1-7.2 (m, 9Harom.), 7.1 (br.s, 1H, NH, exchangeable with D2O), 3.87 (s, 3H, OMe) and 2.9 (s, 3H, Me). Furthermore, the EI-MS of 10 show the correct molecular ion peak at m/z = 383 (100%) which upon loss of acetonitrile molecule afforded the radical cation at m/z = 342 (22.3%).The formation of compounds 8-10 is assumed to proceed via nucleophilic addition of the hydrazino nitrogen nucleophile to the activated carbonyl and thiocarbonyl group via tetrahedral mechanism followed by 1,5-exo-trig cyclization. (Scheme 4)The structure 11b was confirmed by analysis of IR, 1HNMR and mass spectrum which were completely in accord with the assigned structure. (C.f. Exp.)When solution of 5 in dilute hydrochloric acid was stirred with sodium nitrite solution at 0oC yielded the pyridotetrazolo pyrimidine derivative 12 whose structure was deduced from the study of IR and mass spectrum (C.f. Exp.).
Ample evidence for the structure 9 is forthcoming from 1H NMR analysis which completely consistent with the proposed structure (C.f. Exp.). Treatment of compound 5 with freshly distilled acetic anhydride yielded 3-methyl-8-phenyl -6-(4-methoxyphenyl)-1H,2H-pyrido[2,3-d]1,2,4-triazolol[4,3-a]pyrimidin-5-one 10 whose structure was deduced from the correct analytical and spectroscopic data. The IR spectrum of 10 displayed one absorption band for the carbonyl group at 1699 cm-1 and 1H NMR spectrum [DMSO-d6] revealed signals at δ (ppm) 8.6 (s, 1H, C7-H), 8.1-7.2 (m, 9Harom.), 7.1 (br.s, 1H, NH, exchangeable with D2O), 3.87 (s, 3H, OMe) and 2.9 (s, 3H, Me). Furthermore, the EI-MS of 10 show the correct molecular ion peak at m/z = 383 (100%) which upon loss of acetonitrile molecule afforded the radical cation at m/z = 342 (22.3%).The formation of compounds 8-10 is assumed to proceed via nucleophilic addition of the hydrazino nitrogen nucleophile to the activated carbonyl and thiocarbonyl group via tetrahedral mechanism followed by 1,5-exo-trig cyclization. (Scheme 4)The structure 11b was confirmed by analysis of IR, 1HNMR and mass spectrum which were completely in accord with the assigned structure. (C.f. Exp.)When solution of 5 in dilute hydrochloric acid was stirred with sodium nitrite solution at 0oC yielded the pyridotetrazolo pyrimidine derivative 12 whose structure was deduced from the study of IR and mass spectrum (C.f. Exp.).3. Biological Investigations
3.1. Antimicrobial activity
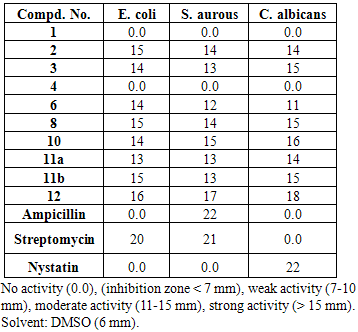
- The antimicrobial screening of all the synthesized compounds was done using the agar diffusion assay. This screening was performed against the Gram-negative bacteria, Escherichia coli ATCC 10536 and Gram-positive bacteria, Staphylococcus aurous ATCC 06538 in addition to the pathogenic fungi Candida albicans ATCC 1023 and Aspergilus flavus. A moderate activity was observed with compounds 2, 3, 6, 8, 10, 11a, 11b and 12 which proved to possess marked activity against E. coli, S. aurous, and C. albicans. The inhibitory concentration was determined for each of the active compounds along with Ampicillin, Streptomycin, and Nystatin as positive controls. No activity was detected for all the synthesized compounds towards Aspergilus flavus. Results are shown in Table 1.
|
4. Experimental
- All melting points were taken on Griffin and Geory melting point apparatus and are uncorrected. IR spectra were recorded on Pye Unicam SP 1200 spectrophotometer using KBr Wafer technique. 1H-NMR spectra were determined on a Varian Gemini 300 MHz using TMS as internal standard (chemical shifts in δ-scale). The mass spectra were determined using HP model MS-5988 at electron energy 70 eV. Elemental analyses were carried out at the microanalytical unit, faculty of science, Cairo University by using Perkin-Elmer 2400 CHN elemental analyzer.5-(4-Methoxyphenyl)-7-phenyl-2-thioxo-2,3-dihydropyrido [2,3-d]pyrimidin-4(3H)-one 1A mixture of 1 (1.43 g, 0.01 mol) and 2 (2.4 g, 0.01 mol) was refluxed in (50 ml) absolute ethanol with (2 ml) piperidine for 4 hours. The reaction mixture was allowed to cool, and then poured into ice hydrochloric acid. The yellow product deposited was filtered off, dried and then recrystallized from dioxane to give 1 as yellow crystals, m.p.>300oC (yield 66%). IR (ν cm-1): 3398, 3293(2NH), 1697 (C=O), 1173 (C=S). 1HNMR (DMSO-d6): δ 13 (s, 1H, NH, broad exchangeable with D2O), 12.3 (s, 1H, NH, broad exchangeable with D2O), 8.4 (s, 1H, C6-H), 8.29-6.9 (m, 9Harom), 3.7 (s, 3H, OMe). MS m/z (%): 361 (M+., 100), 360 (93), 301 (8), 318 (8). Anal. Calcd. For C20H15N3O2S (361): C, 66.48; H, 4.16; N, 11.63; S, 8.86, Found: C, 66.62; H, 3.95; N, 11.42; S, 9.02.2-Carboxymethylthio-5-(4-methoxyphenyl)-7-phenyl pyrido[2,3-d]-pyrimidin-4(3H)-one 2Compound 1 (1.8 g, 0.005 mol) was refluxed with ethyl chloroacetate (0.74 g, 0.006 mol) in n-butanol in the presence of anhydrous sodium acetate (2.7 g) for 10 hours. The separated solid was washed with warm water several times and then filtered off, dried and recrystallized from ethanol to give 2 as brown crystals; m.p. 210-211oC (yield 65%). IR (ν cm-1): 3452 (broad -OH), 1701 (C=O, acid), 1687(C=O, imide). 1HNMR (DMSO-d6): δ 12.6 (s, 1H, NH, broad exchangeable with D2O), 11.3 (br.s, 1H, COOH, exchangeable with D2O), 8.7(s, 1H, C6-H), 8.3-7.1 (m, 9Harom), 3.9 (s, 2H, CH2COOH), 3.9 (s, 3H, OMe). MS m/z (%): 419 (M+., 82), 401 (M+-H2O, 17), 391 (56), 77 (74). Anal. Calcd. For C22H17N3O4S (419): C, 63.01; H, 4.06; N, 10.02; S, 7.63, Found: C, 63.23; H, 3.90; N, 10.25; S, 7.45. 3,5-Dioxo-6-(4-methoxyphenyl)-8-phenyl-2H,3H-pyrido 2,3-d]thiazolo[3,2-a] pyrimidine 3Method 1Compound 2 (1 g, 0.002 mol) was refluxed with freshly distilled acetic anhydride (10 ml) for 3 hours. The reaction mixture was allowed to cool, and then poured into ice/water and left overnight in the refrigerator. The dark green precipitate was filtered off, washed several times with water, dried and then recrystallized from methanol to give 3 as pale green crystals m.p. 250-251oC (yield 49%). IR (ν cm-1): 1670, 1682 (2C=O). 1HNMR (DMSO-d6): δ 8.7(s, 1H, C7-H), 8.3-7.7 (m, 9Harom), 4.1 (s, 2H, SCH2CO), 3.9 (s, 3H, OMe). MS m/z (%): 401 (M+., 12), 360 (100). Anal. Calcd. For C22H15N3O3S (401): C, 65.84; H, 3.74; N, 10.47; S, 7.98, Found: C, 66.00; H, 3.61; N, 10.28; S, 8.11. Method 2To a stirred solution of compound 1 (1.44 g, 0.004 mol) in 20 ml pyridine, the chloroacetyl chloride (0.44 ml, 0.004 mol) was added dropwise over a period of 10 min., then the reaction mixture was refluxed on water bath for 1 hour. The reaction mixture was poured into ice cold acetic acid and the solid deposited was filtered off, washed several times with light petroleum ether, dried and then recrysytallized from methanol to give 3 (yield 72%).2-Ethylthio-5-(4-methoxyphenyl)-7-phenylpyrido[2,3-d] pyrimidin-4(3H)-one 4A mixture of 1 (3 g, 0.008 mol) and ethyl iodide (0.7 ml, 0.008 mol) was refluxed in (50 ml) absolute ethanol in the presence of anhydrous sodium acetate (2 g) for 3 hours The organic layer was separated and slow evaporates to give green solid which was washed with warm water and then filtered off, dried and then recrystallized from ethanol to give 4 as green crystals m.p. 310-311oC (yield 64%). IR (ν cm-1): 3127 (NH), 1680 (C=O). 1HNMR (DMSO-d6): δ 12.4(s, 1H, NH, exchangeable with D2O), 8.6 (s, 1H, C6-H), 8.3-7.1(m, 9Harom), 3.9(s, 3H, OMe), 3.15 (q, 2H, SCH2CH3), 1.36 (t, 3H, SCH2CH3). MS m/z (%): 389 (M+., 18), 361 (100), 360 (77), 344 (18), 302 (7), 181 (17). Anal. Calcd. For C22H19N3O2S (389): C, 67.87; H, 4.88; N, 10.80; S, 8.23, Found: C, 67.73; H, 4.80; N, 10.66; S, 8.12.2-Hydrazinyl-5-(4-methoxyphenyl)-7-phenylpyrido[2,3-d] pyrimidin-4(3H)-one 5Method 1Compound 1 (3.6 g, 0.01 mol) was refluxed with hydrazine hydrate (80%) (1.25 ml, 0.025 mol) in ethanol (20 ml) for 2 hours. The solid deposited on hot was filtered off, dried and then recrystallized from dimethylformamide to give 5 as green crystals, m.p. 275-277oC (yield 39.6%).Method 2Compound 4 (1 g, 0.0025 mol) was refluxed with excess hydrazine hydrate (80%) in ethanol (20 ml) for 2 hours. The solid deposited during the reflux was filtered off, dried and recrystallized from dimethylformamide to give 5 (yield 56.6%); IR (ν cm-1): 3350, 3196 (NH2), 1662 (C=O). 1HNMR (DMSO-d6): δ 10.3 (br.s, 1H, NH, exchangeable with D2O), 8.6 (s,1H, C6-H), 8.3 (br.s, 3H, NHNH2, exchangeable with D2O), 8.1-7.1 (m, 9Harom), 3.9 (s, 3H, OMe). MS m/z (%): 359 (M+., 100), 358 (40), 329 (31), 300 (7). Anal. Calcd. For C20H17N5O2 (359): C, 66.85; H, 4.74; N, 19.50, Found: C, 66.68; H, 4.71; N, 19.32.6-(4-Methoxyphenyl)-8-phenylpyrido[2,3-d]-1,2,4-triazolo [4,3-a]-pyrimidin-5-one 6Compound 5 (1 g, 0.003 mol) was refluxed with triethylorthformate (10 ml) in the presences of acetic anhydride (5 ml) for 16 hours. The reaction mixture was allowed to cool, and the obtained solid was filtered off, washed with petroleum ether 80-100oC, dried and recrystallized from ethanol to give 6 as buff crystals, m.p. 300oC (yield 80%). IR (ν cm-1): 3120 (NH), 1714 (C=O). 1HNMR (DMSO-d6): δ 8.7 (s, 1H, C7-H), 8.4-7.2 (m, 9Harom), 7.0 (br.s, 1H, NH, exchangeable with D2O), 6.1 (s, 1H, C3-H), 3.9 (s, 3H, OMe). MS m/z (%): 369 (M+., 100), 368 (67), 342 (18), 327 (12). Anal. Calcd. For C21H15N5O2 (369): C, 68.29; H, 4.04; N, 18.97, Found: C, 68.15; H, 4.29; N, 18.78.6-(4-Methoxyphenyl)-8-phenyl-1H,2H-pyrido[2,3-d]-1,2,4-triazolo[4,3-a]-pyrimidin-3,5-dione 8A mixture of 5 (1 g, 0.003 mol), ethyl chloroformate (5 ml) and pyridine (20 ml) was refluxed for 6 hours. The reaction mixture was poured into water to give light brown solid which filtered off, dried and then recrystallized from ethanol to give 8 as yellow crystals, m.p. 228-230oC (yield 50%). IR (ν cm-1): 3256 (NH), 1731, 1706 (2C=O). 1HNMR (DMSO-d6): δ 8.6 (s, 1H, C7-H), 8.3-7.0 (m, 9Harom.), 6.0 (br.s, 2H, 2NH, exchangeable with D2O), 3.9 (s, 3H, OMe). MS m/z (%): 385 (M+., 100), 342 (15), 328 (17), 286 (20). Anal. Calcd. For C21H15N5O3 (385): C, 65.45; H, 3.90; N, 18.18, Found: C, 65.48; H, 4.01; N, 18.33.6-(4-Methoxyphenyl)-8-phenyl-3-thioxo-1H,2H-pyrido [2,3-d]-1,2,4-triazolo[4,3-a]-pyrimidin-5-one 9A mixture of 5 (0.6 g, 0.0017 mol) and phenyl isothiocyanate (0.4 ml, 0.003 mol) was refluxed in pyridine (15 ml) for 8 hours. The reaction mixture was poured onto ice/hydrochloric acid. The solid obtained was filtered off, dried and then recrystallized from benzene to give 9 as pink crystals, m.p. 110-112oC (yield 35%). IR (ν cm-1): 3381 (NH, broad), 1676 (C=O), 1246 (C=S). 1HNMR (DMSO-d6): δ 8.7(s, 1H, C7-H), 8.1-7.2 (m, 9Harom.), 6.0 (br.s, 2H, 2NH, exchangeable with D2O), 3.9 (s, 3H, OMe). MS m/z (%): 369 (M+.-S, 100). Anal. Calcd. For C21H15N5O2S (401): C, 62.84; H, 3.74; N, 17.46; S, 7.98, Found: C, 62.70; H, 3.91; N, 17.25; S, 8.18.3-Methyl-6-(4-methoxyphenyl)-8-phenylpyrido[2,3-d]-1,2,4-triazolo[4,3-a]pyrimidin-5(1H)-one 10Compound 5 (1 g, 0.003 mol) was refluxed in acetic anhydride (10 ml) for 6 hours. The reaction mixture was poured onto ice/water. The yellow solid obtained was filtered off, dried and recrysytallized from methanol to give 10 as yellow crystals, m.p. 260-261oC (yield 72%). IR (ν cm-1): 3317 (NH), 1699 (C=O). 1HNMR (DMSO-d6): δ 8.6 (s, 1H, C7-H), 8.1-7.2 (m, 9Harom), 7.1 (br.s, 1H, NH, exchangeable with D2O), 3.8 (s, 3H, OMe), 2.3 (s, 3H, C3-Me). MS m/z (%): 383 (M+., 100), 382 (54), 368 (19), 342 (22). Anal. Calcd. For C22H17N5O2 (383): C, 68.93; H, 4.44; N, 18.28, Found: C, 69.11; H, 4.62; N, 18.08.10-Phenyl-3-Methyl-8-(4-methoxyphenyl)-5-(substituted)-7-oxo-1H-pyrido[2,3-d]pyrimido[2,1-c][1,2,4]triazepine 11a, bA mixture of 5 (1 g, 0.003 mol) with acetyl acetone (0.3 ml, 0.003 mol) and/or benzoyl acetone (0.48 g, 0.003 mol) in dimethylformamide (30 ml) was refluxed for 8 hours. The reaction mixture poured onto water. The organic layer was extracted with methylene chloride, dried over magnesium sulfate and slow evaporated to give 11a and 11b, respectively. 11a: recrystallized from toluene as buff crystals, m.p. 150-152oC (yield 69%). IR (νcm-1): 3312 (NH), 1698 (C=O). 1HNMR (DMSO-d6): δ 10.4 (s, 1H, NH, exchangeable with D2O), 8.2-6.9 (m, 10Harom+C9-H), 6.1 (s, 1H, C4-H), 3.8 (s, 3H, OMe), 2.9 (s, 3H, Me), 2.27 (s, 3H, C5-Me). MS m/z (%): 423 (M+., 98), 422 (100), 408 (27), 329 (18). Anal. Calcd. For C25H21N5O2 (423): C, 70.92; H, 4.96; N, 16.55, Found: C, 70.80; H, 4.92; N, 16.80.11b: recrystallized from ethanol as green crystals, m.p. 250-251oC (yield 58%). IR (ν cm-1): 3368 (NH), 1697 (C=O). 1HNMR (DMSO-d6): δ 10.3 (s, 1H, NH, exchangeable with D2O), 8.2-7.3 (m, 14Harom), 6.9 (s, 1H, C9-H), 6.1 (s, 1H, C4-H), 3.6 (s, 3H, Me), 3.8 (s, 3H, OMe). MS m/z (%): 485 (M+., 59), 383 (100), 329 (61), 328 (65), 314 (30), 300 (24). Anal. Calcd. For C30H23N5O2 (485): C, 74.22; H, 4.74; N, 14.43, Found: C, 74.20; H, 4.70; N, 14.29.6-(4-Methoxyphenyl)-8-phenylpyrido[2,3-d]tetrazolo[1,5-a]pyrimidin-5(1H)-one 12Compound 5 (1 g, 0.003 mol) was dissolved in (10 ml) concentrated hydrochloric acid and then (7 ml) of sodium nitrite (10%) was added dropwise with stirring over a period of 2 hours at 0oC. The obtained yellow solid was filtered off, dried and then recrystallized from ethanol to give 12 as yellow crystals, m.p. 230-232oC (yield 39%). IR (ν cm-1): 3384 (NH), 1700 (C=O). 1HNMR (DMSO-d6): δ 8.5 (s, 1H, C7-H), 7.8-7.0 (m, 9Harom), 5.4 (br.s, 1H, NH, exchangeable with D2O) 3.9 (s, 3H, OMe). MS m/z (%): 370 (M+., 37), 369 (100), 344 (42), 329 (22). Anal. Calcd. For C20H14N6O2 (370): C, 64.86; H, 3.78; N, 22.70, Found: C, 65.03; H, 3.70; N, 22.53.