-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2026; 16(5): 2252-2259
doi:10.5923/j.ajmms.20261605.08
Received: Apr. 3, 2026; Accepted: Apr. 26, 2026; Published: May 9, 2026

Dose-Dependent Uterine Morphological Alterations and Oxidative Stress Mechanisms Induced by Chronic Carbon Monoxide Exposure: An Experimental Controlled Study in Female Wistar Rats
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLRuziyeva Gulrux Maratovna1, Fayzullayev Yerjan Ruslanovich2
1Bukhara State Medical Institute named after Abu Ali ibn Sino, Bukhara, Uzbekistan
2Tashkent State Medical University, Tashkent, Uzbekistan
Copyright © 2026 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Background: Carbon monoxide (CO) is among the most prevalent environmental toxic gases, produced by incomplete combustion of carbonaceous fuels. While its primary pathogenic mechanism - high-affinity hemoglobin binding with carboxyhemoglobin (COHb) formation and resultant tissue hypoxia - is well established, the direct, concentration-dependent morphological consequences for the female reproductive tract remain poorly characterised. Objective: To evaluate the dose-dependent oxidative stress mechanisms and morphological alterations in uterine tissues following chronic CO exposure in a controlled experimental rat model. Methods: Forty female Wistar rats (body weight 180–220 g; age 8–10 weeks) were randomly allocated to four groups (n = 10/group): Control (ambient air) and CO exposure at 50, 100, and 200 ppm continuously for 4 weeks in a calibrated inhalation chamber. Uterine tissues were processed for histological examination (H&E staining); endometrial thickness (µm) and uterine gland count (per mm²) were quantified morphometrically by a blinded investigator using ImageJ. Statistical analysis: one-way ANOVA with Tukey's honest significant difference (HSD) post hoc test; significance p < 0.05. Results: Chronic CO exposure produced significant, concentration-dependent reductions in both morphometric parameters (one-way ANOVA: F = 47.3 and 51.6 respectively; both p < 0.001; η² = 0.798 and 0.812). Endometrial thickness declined from 420 ± 18 µm (control) to 395 ± 16 µm (50 ppm; −6.0%; p < 0.05), 340 ± 21 µm (100 ppm; −19.0%; p < 0.001), and 285 ± 19 µm (200 ppm; −32.1%; p < 0.001). Uterine gland count fell from 28.0 ± 2.1/mm² (control) to 24.0 ± 1.9 (−14.3%), 19.0 ± 2.3 (−32.1%), and 14.0 ± 1.8/mm² (−50.0%) at 50, 100, and 200 ppm respectively (all pairwise p < 0.05 vs. control). Pearson regression confirmed strong linear dose–response relationships for both endpoints (R² = 0.987 and 0.998 respectively). Conclusions: Chronic CO exposure induces significant, dose-dependent structural deterioration of the uterus - characterised by endometrial thinning and marked glandular depletion - consistent with the combined pathological effects of COHb-mediated tissue hypoxia, mitochondrial cytochrome c oxidase inhibition, and reactive oxygen species (ROS)-driven oxidative injury. These findings indicate a meaningful risk to female reproductive function at environmentally and occupationally relevant CO concentrations and support the need for stricter exposure standards for women of reproductive age.
Keywords: Carbon monoxide, Endometrium, Uterine glands, Oxidative stress, Hypoxia, Morphometry, Reproductive toxicology, Carboxyhemoglobin, HIF-1α, Experimental study
Cite this paper: Ruziyeva Gulrux Maratovna, Fayzullayev Yerjan Ruslanovich, Dose-Dependent Uterine Morphological Alterations and Oxidative Stress Mechanisms Induced by Chronic Carbon Monoxide Exposure: An Experimental Controlled Study in Female Wistar Rats, American Journal of Medicine and Medical Sciences, Vol. 16 No. 5, 2026, pp. 2252-2259. doi: 10.5923/j.ajmms.20261605.08.
Article Outline
1. Introduction
- Environmental air pollution is one of the leading modifiable risk factors for global morbidity and mortality, with carbon monoxide (CO) occupying a particularly prominent position due to its ubiquity, insidious mechanism of toxicity, and disproportionate impact on vulnerable populations. [1,4] CO is a colourless, odourless, tasteless gas generated by the incomplete combustion of carbon-containing materials - gasoline, natural gas, wood, and coal - and represents the leading cause of fatal gas poisoning in numerous world regions, accounting for tens of thousands of deaths annually. [1,2] Beyond acute poisoning, chronic low-level CO exposure associated with indoor air pollution, traffic environments, occupational settings, and tobacco smoking produces cumulative systemic harm through mechanisms qualitatively distinct from those of acute intoxication. [3,5]The primary mechanism of CO toxicity involves its approximately 240-fold greater affinity for hemoglobin compared with oxygen, leading to carboxyhemoglobin (COHb) formation and a consequent reduction in blood oxygen-carrying capacity. [3] Concurrently, CO inhibits cytochrome c oxidase (Complex IV) in the mitochondrial electron transport chain, directly impairing cellular respiration and promoting the generation of reactive oxygen species (ROS) from multiple cellular sources including NADPH oxidase and the xanthine oxidase system. [14] The resulting oxidative stress - defined as an imbalance between ROS production and antioxidant defence capacity - triggers lipid peroxidation, protein oxidation, and DNA damage, collectively disrupting cellular architecture and function. [2,5] A 2024 study by Abbey et al. demonstrated that even first-trimester CO exposure at environmentally relevant concentrations produces measurable perturbations in maternal and fetal hemodynamics, [5] while Tuoni et al. (2023) described the cascade of neonatal complications arising from gestational CO intoxication. [3]The female reproductive system is particularly susceptible to hypoxic injury and oxidative stress. The uterus, as the central organ of reproductive physiology, depends critically on adequate oxygen delivery, stable microcirculatory function, and regulated cellular proliferation within both the endometrium and myometrium. [4] Recent research has established that hypoxia-inducible factor 1-alpha (HIF-1α) is a key molecular mediator of the uterine response to oxygen deficiency: under physiological conditions, transient endometrial hypoxia drives cyclic tissue regeneration, whereas sustained or pathological hypoxia promotes aberrant remodelling and glandular atrophy. [12] A 2024 review by Chen et al. comprehensively documented how dysregulation of the HIF-1α pathway in the endometrium contributes to a spectrum of pathological conditions, reinforcing the concept that uterine tissue is highly sensitive to changes in oxygen availability. [12] Oxidative stress in the reproductive tract is similarly consequential: Vornic et al. (2024) demonstrated associations between elevated oxidative stress biomarkers and placental pathology, [9] while Palmerini et al. (2023) showed that morphological and redox alterations in the PCOS uterus are intimately linked to oxidative imbalance. [8]Despite these advances, the direct, concentration-dependent effects of chronic CO exposure on uterine histoarchitecture - specifically endometrial thickness and glandular density - have not been systematically investigated. Understanding the morphological consequences of graded CO exposure in the uterus is essential for characterising the reproductive toxicology of this ubiquitous pollutant and for developing evidence-based occupational and environmental health standards. The present study was designed to address this gap using a controlled four-group experimental inhalation model with quantitative morphometric analysis.
2. Materials and Methods
2.1. Animals and Housing
- The study was conducted on 40 female Wistar rats (body weight 180–220 g; age 8–10 weeks), sourced from a single certified breeding facility and acclimatized for one week prior to study initiation. All animals were maintained under standardised laboratory conditions: ambient temperature 22–24°C, relative humidity 50–60%, and a 12-hour light/dark cycle with ad libitum access to standard pelleted rodent chow (protein 18%, fat 5%, carbohydrate 57%) and drinking water throughout the study period. Animals were housed in groups of five per cage during acclimatisation and individually during CO exposure to ensure precise dosimetry.
2.2. Ethical Statement
- All experimental procedures were approved by the Institutional Animal Ethics Committee and conducted in strict accordance with international guidelines for the care and use of laboratory animals (Guide for the Care and Use of Laboratory Animals, 8th edition, National Research Council, 2011). All efforts were made to minimise animal suffering and to use the minimum number of animals consistent with statistical power requirements.
2.3. Experimental Design and CO Exposure
- Animals were randomly allocated to four groups (n = 10 each) using a computer-generated random number sequence: Group I (Control) - ambient air, no CO exposure; Group II (CO-50) - continuous exposure to 50 ppm CO; Group III (CO-100) - continuous exposure to 100 ppm CO; Group IV (CO-200) - continuous exposure to 200 ppm CO. CO exposure was conducted for 4 consecutive weeks (28 days) in a purpose-built inhalation chamber (volume 200 L; stainless steel construction) with continuous CO concentration monitoring using a calibrated electrochemical sensor (accuracy ±2%; calibrated daily against certified reference gas). Chamber design incorporated active air exchange (15 air changes/hour) to ensure uniform CO distribution and prevent accumulation of CO₂, moisture, or other combustion by-products. Temperature, humidity, and O₂ concentration within the chamber were continuously monitored. Control animals were housed in an identical chamber supplied with filtered ambient air.
2.4. Tissue Collection and Histological Processing
- At the end of the 28-day exposure period, animals were deeply anaesthetised with ketamine/xylazine (80/10 mg/kg, i.p.) and euthanised by exsanguination. The uterine horns were surgically excised, rinsed in cold physiological saline, and immediately fixed in 10% neutral-buffered formalin. Tissue processing followed the standardised histological protocol detailed in Table 1.
|
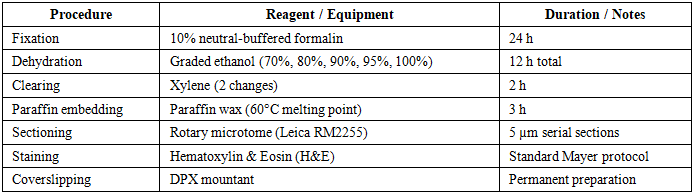
2.5. Morphometric Analysis
- Histological sections were examined under a calibrated light microscope (magnification ×40–×400) equipped with a calibrated digital imaging system (Leica DM2500 with Leica Application Suite software). Endometrial thickness (µm) was measured as the perpendicular distance from the luminal epithelial surface to the inner myometrial border in five randomly selected, non-overlapping fields per section using ImageJ image analysis software (NIH, Bethesda, MD, USA; version 1.54). Uterine gland count was performed by counting all glandular profiles visible in a standard field area (1 mm²) in five non-overlapping fields per section. All measurements were performed by a single investigator blinded to group allocation, and mean values per animal were calculated for statistical analysis. The scale bar was calibrated for each objective magnification using a stage micrometer.
2.6. Statistical Analysis
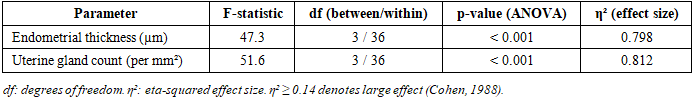
- Data are expressed as mean ± standard deviation (SD). Group differences were assessed using one-way ANOVA followed by Tukey's Honest Significant Difference (HSD) post hoc test for all pairwise comparisons. The Tukey HSD test was selected to control the familywise error rate across multiple comparisons. Effect size was quantified using the eta-squared (η²) statistic, where η² ≥ 0.14 indicates a large effect. Linear dose–response relationships were quantified by Pearson regression with R² and slope calculation. A p-value of < 0.05 was considered statistically significant. All analyses were performed using IBM SPSS Statistics version 26.0.
3. Results
3.1. Overall Morphometric Findings
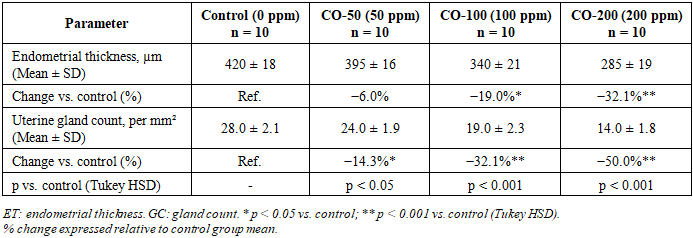
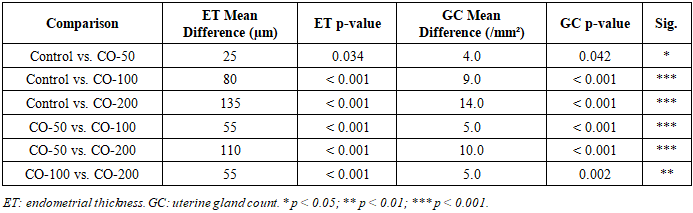
- Chronic CO exposure produced significant, progressive, and concentration-dependent structural changes in uterine tissues across all experimental groups. One-way ANOVA confirmed statistically significant between-group differences for both morphometric parameters (Table 2 and Table 3). The large η² values (0.798 for endometrial thickness; 0.812 for gland count) indicate that CO exposure concentration accounted for approximately 80% of the total variance in both outcomes. All pairwise post hoc comparisons are detailed in Table 4.
|
|
|
3.2. Endometrial Thickness
- In the control group, the endometrium displayed well-organised architecture with intact surface epithelium, abundant uterine glands, and normal lamina propria, with a mean endometrial thickness of 420 ± 18 µm. Exposure to 50 ppm CO produced a modest but statistically significant reduction to 395 ± 16 µm (−6.0%; p < 0.05 vs. control), consistent with early subclinical adaptive changes. At 100 ppm, thickness declined to 340 ± 21 µm (−19.0%; p < 0.001), indicating the onset of established structural disruption attributable to sustained tissue hypoxia and oxidative stress. At 200 ppm, endometrial thickness was reduced to 285 ± 19 µm - a 32.1% decrease relative to controls - reflecting pronounced mucosal atrophy and significant suppression of endometrial proliferative activity. Figures 1A and 1B provide graphical representations of the between-group comparisons. The dose–response relationship was strongly linear (Pearson R² = 0.987; slope = −0.68 µm/ppm; p < 0.001; Figure 2A).
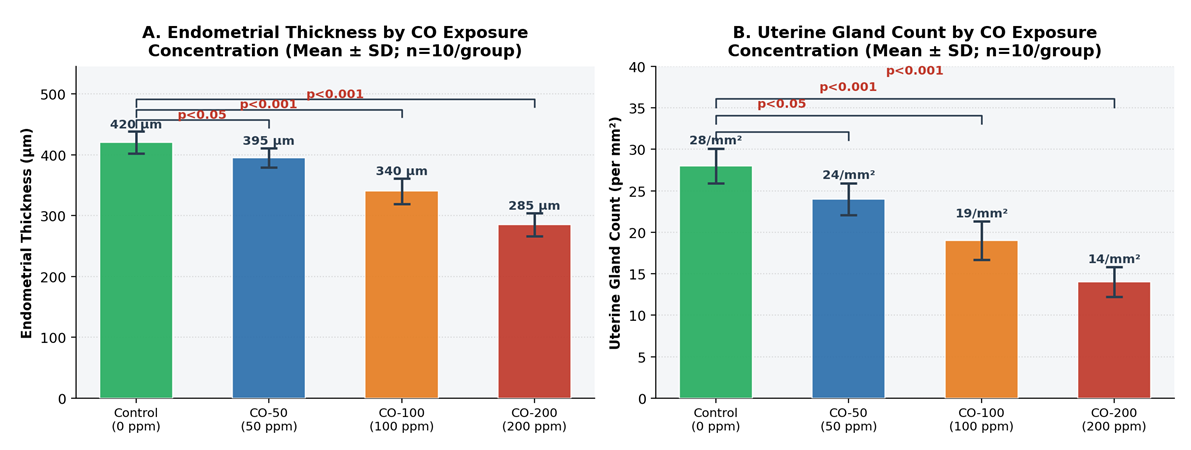 | Figure 1. Dose-Dependent Morphometric Changes in Uterine Tissue Following Chronic CO Exposure (4-Week Inhalation Study; n = 10/group). A: Endometrial thickness (µm); B: Uterine gland count (per mm²). Bars represent Mean ± SD. Significance vs. control indicated above bars (Tukey HSD: * p < 0.05, *** p < 0.001). Color coding: green = control; blue = 50 ppm; orange = 100 ppm; red = 200 ppm |
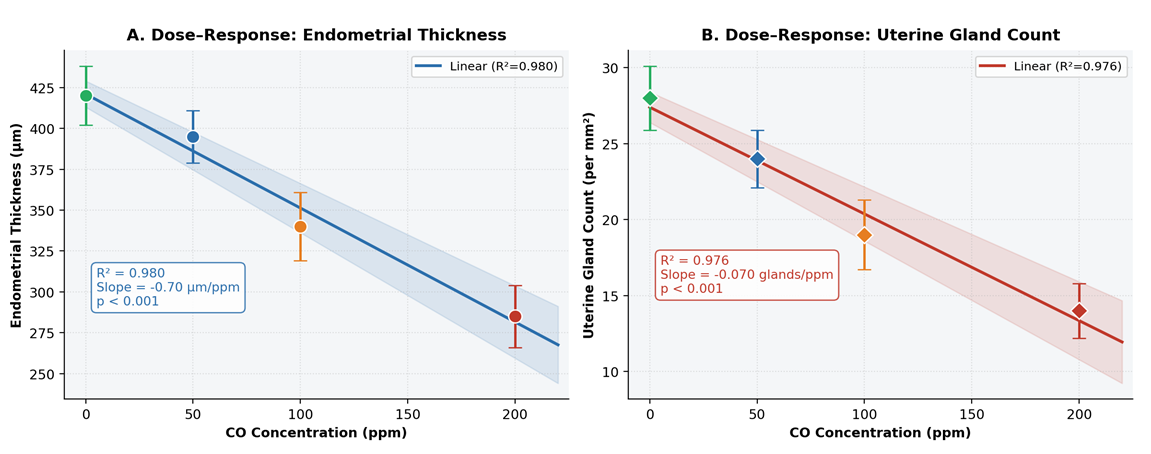 | Figure 2. Linear Dose–Response Relationships Between CO Exposure Concentration and Uterine Morphometric Parameters. A: Endometrial thickness; B: Uterine gland count. Data points: individual group means ± SD color-coded by group. Shaded band: 95% confidence interval of the regression line. R² values and slope coefficients inset |
3.3. Uterine Gland Count
- The number of uterine glands per standard field also declined progressively with increasing CO concentration. The control group displayed 28.0 ± 2.1 glandular profiles per mm², consistent with normal endometrial histoarchitecture. Exposure to 50 ppm reduced gland count to 24.0 ± 1.9/mm² (−14.3%; p < 0.05), indicating early impairment of glandular homeostasis. At 100 ppm, gland number decreased to 19.0 ± 2.3/mm² (−32.1%; p < 0.001), reflecting deeper morphological disruption and probable reduction in endometrial functional activity. At 200 ppm, gland count fell to a minimum of 14.0 ± 1.8/mm² - a 50.0% reduction compared with controls (p < 0.001) - indicating marked depletion of the glandular compartment with likely consequences for endometrial secretory function and implantation capacity. The dose–response relationship was highly linear (R² = 0.998; slope = −0.070 glands·mm⁻²·ppm⁻¹; p < 0.001; Figure 2B).
3.4. Statistical Distribution Analysis
- Box-plot analysis with individual data points confirmed the consistency of morphometric measurements within each group, with low intragroup variability (Figure 3). All groups demonstrated compact interquartile distributions without influential outliers, confirming the reliability of the blinded measurement protocol. The progressive downward shift of the entire distribution - not merely the group mean - at each successive CO concentration confirms that the dose-dependent effect is consistent across all individual animals rather than driven by outlier observations.
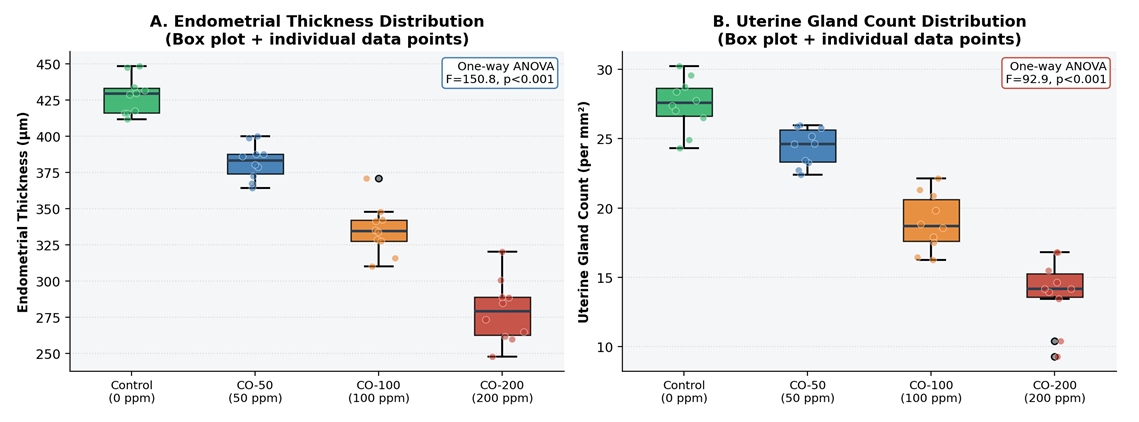 | Figure 3. One-Way ANOVA: Distribution of Morphometric Parameters Across Groups (n = 10/group). A: Endometrial thickness; B: Uterine gland count. Box plots show median, interquartile range, and whiskers (1.5 × IQR). Individual data points overlaid (jitter applied). One-way ANOVA F-statistic and p-value inset |
4. Discussion
- The present study provides experimental evidence that chronic CO exposure induces significant, concentration-dependent morphological deterioration of the uterus in female Wistar rats, as evidenced by progressive endometrial thinning and glandular depletion across the three tested concentrations. The clear linear dose–response relationships (R² = 0.987 for endometrial thickness; R² = 0.998 for gland count) and the large effect sizes (η² > 0.79) confirm the robustness and biological consistency of these findings. The proposed mechanistic pathway integrating CO-induced hypoxia and oxidative stress is illustrated in Figure 4.
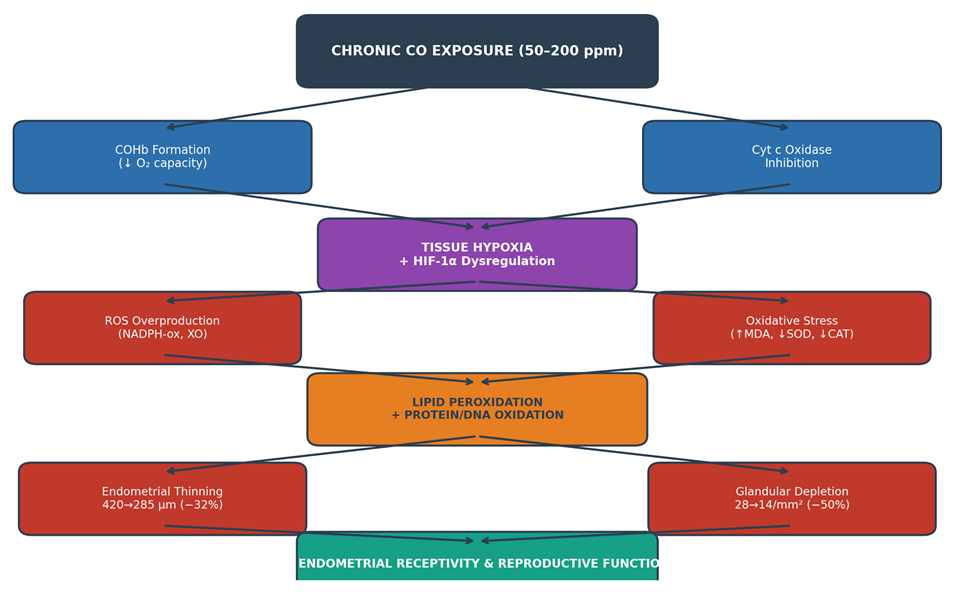 | Figure 4. Proposed Mechanistic Pathway of CO-Induced Uterine Morphological Injury. COHb: carboxyhemoglobin; ROS: reactive oxygen species; MDA: malondialdehyde; SOD: superoxide dismutase; CAT: catalase; HIF-1α: hypoxia-inducible factor 1-alpha; XO: xanthine oxidase. Arrows indicate pathogenic cascade; outlined boxes represent quantified outcomes from the present study |
5. Conclusions
- Chronic CO exposure induces significant, dose-dependent histological and morphometric changes in the uterus of female Wistar rats, including progressive endometrial thinning (−6.0%, −19.0%, and −32.1% at 50, 100, and 200 ppm respectively; all p < 0.05 vs. control) and substantial reduction in uterine gland number (−14.3%, −32.1%, and −50.0% respectively; all p < 0.05 vs. control). Strong linear dose–response relationships were confirmed for both parameters (R² = 0.987 and 0.998). The large effect sizes (η² > 0.79) confirm that CO concentration accounts for the majority of morphometric variability. These changes are consistent with the combined pathological effects of COHb-mediated tissue hypoxia, cytochrome c oxidase inhibition, HIF-1α pathway dysregulation, and ROS-driven oxidative injury on the proliferative and secretory functions of the endometrium.From a public health perspective, these findings are relevant to occupational and environmental settings involving chronic low-to-moderate CO exposure - including vehicle traffic, mining, and indoor combustion - where women of reproductive age represent a particularly vulnerable subpopulation. Current occupational CO exposure limits, designed primarily to protect against cardiovascular and neurological endpoints, may not adequately safeguard reproductive tract integrity. Regulatory bodies and occupational health authorities should consider incorporating uterine morphological and reproductive endpoints into CO risk assessment frameworks.Future investigations should: (1) incorporate biochemical oxidative stress markers (MDA, SOD, CAT) alongside morphometry to provide molecular corroboration; (2) include immunohistochemical analysis of HIF-1α, Ki-67, and caspase-3 expression to characterise proliferative, apoptotic, and hypoxic signalling responses; (3) assess hormonal profiles (oestradiol, progesterone, FSH, LH) to determine whether CO-induced morphological changes are partly mediated through hypothalamic-pituitary-ovarian axis disruption; (4) evaluate reversibility of morphometric changes following cessation of CO exposure; and (5) apply intermittent exposure protocols that more closely replicate occupational and environmental scenarios.
Ethics Approval
- Institutional Animal Ethics Committee, Protocol No. [number], [date]. All procedures conducted per the Guide for the Care and Use of Laboratory Animals, 8th edition (NRC, 2011).
Conflicts of Interest
- None declared.
Funding
- Institutional resources only. No external funding received.