-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2026; 16(2): 670-675
doi:10.5923/j.ajmms.20261602.58
Received: Nov. 29, 2025; Accepted: Dec. 22, 2025; Published: Feb. 25, 2026

Impact of Complex Therapeutic Agents on Liver Protective Mechanisms: A Literature Analysis
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLSaidov Saidamir, Pulatova Dilfuza
Institute of Pharmaceutical Education and Research, Yunusabad/19, Yunus ota, Tashkent, Uzbekistan
Correspondence to: Saidov Saidamir, Institute of Pharmaceutical Education and Research, Yunusabad/19, Yunus ota, Tashkent, Uzbekistan.
| Email: |  |
Copyright © 2026 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

The liver, as the central organ for xenobiotic metabolism, is perpetually exposed to the potential hepatotoxic effects of pharmacotherapeutic agents. While single-drug-induced liver injury (DILI) is well-documented, the landscape of modern medicine is increasingly dominated by polypharmacy and complex therapeutic regimens for multimorbid patients. This presents a unique challenge: the unpredictable, synergistic, or antagonistic interactions of multiple drugs on the liver's intrinsic protective mechanisms. This literature analysis aims to synthesize current scientific knowledge on how complex therapeutic agents—defined as multi-drug regimens, fixed-dose combinations, and prodrugs requiring hepatic activation—impact key hepatic protective systems, including antioxidant pathways (GSH, Nrf2), drug-metabolizing enzymes (CYPs, UGTs), mitochondrial integrity, and hepatobiliary transporters. A systematic search of PubMed, Scopus, and Web of Science databases was conducted for literature published between 2000 and 2024. Our analysis reveals that complex regimens, particularly those involving drugs with narrow therapeutic windows (e.g., antiretrovirals, antituberculars, chemotherapeutics), significantly amplify oxidative stress, deplete hepatic glutathione stores by up to 60% in experimental models, and induce mitochondrial permeability transition. Furthermore, pharmacokinetic interactions at the level of CYP450 isoenzymes and efflux transporters (e.g., P-glycoprotein, BSEP) can lead to altered drug exposure and potentiate cholestatic or hepatocellular injury. The Nrf2-Keap1 pathway emerges as a critical but often overwhelmed defense node. This analysis concludes that a paradigm shift from single-drug toxicology to a systems-level understanding of multi-drug interactions on hepatic cytoprotective networks is imperative. Future research must focus on predictive in vitro and in silico models to stratify the hepatotoxic risk of complex therapeutic regimens, thereby guiding safer clinical practice and the development of targeted hepatoprotective adjuvants.
Keywords: Drug-Induced Liver Injury (DILI), Polypharmacy, Hepatoprotection, Oxidative Stress, Cytochrome P450, Mitochondrial Dysfunction, Nrf2 Pathway, Pharmacokinetic Interactions
Cite this paper: Saidov Saidamir, Pulatova Dilfuza, Impact of Complex Therapeutic Agents on Liver Protective Mechanisms: A Literature Analysis, American Journal of Medicine and Medical Sciences, Vol. 16 No. 2, 2026, pp. 670-675. doi: 10.5923/j.ajmms.20261602.58.
Article Outline
1. Introduction
- The liver is a metabolic powerhouse and the primary site for the biotransformation and detoxification of a vast array of endogenous and exogenous compounds, including pharmaceutical agents. This vital function is executed by a sophisticated array of enzymatic systems and cellular mechanisms, collectively termed hepatic protective mechanisms. These include Phase I (e.g., Cytochrome P450 enzymes) and Phase II (e.g., UDP-glucuronosyltransferases, glutathione S-transferases) metabolism, antioxidant defense systems (e.g., glutathione, superoxide dismutase), efficient export via biliary transporters (e.g., BSEP, MRP2), and robust cellular repair processes [3]. However, this very role as a metabolic clearinghouse renders the liver exceptionally vulnerable to injury from the drugs it processes.Drug-induced liver injury (DILI) is a major cause of drug attrition during development, post-marketing warnings, and withdrawal of medications from the clinical arena [2]. Historically, research has predominantly focused on the hepatotoxic potential of single chemical entities. Seminal work by researchers like Hyman J. Zimmerman established the foundational "Hy's Law," linking drug-related hepatocellular jaundice to serious outcomes [7,11]. The pioneering investigations of Nelson and colleagues on the cytochrome P450 superfamily laid the groundwork for understanding metabolic activation and bioactivation of drugs to reactive metabolites, a key concept in intrinsic DILI [4].In the 21st century, the clinical reality has shifted dramatically towards polypharmacy, defined as the concurrent use of five or more medications, a practice particularly prevalent among elderly and chronically ill patients [6]. This paradigm introduces a layer of complexity that is not fully captured by single-agent toxicology studies. The administration of complex therapeutic agents—encompassing multi-drug regimens (e.g., HAART for HIV, RIPE therapy for tuberculosis), fixed-dose combinations (e.g., amoxicillin/clavulanate), and prodrugs requiring hepatic activation—creates a dynamic and interactive milieu within the hepatocyte. The potential for drug-drug interactions (DDIs) at the level of metabolism and transport can lead to unpredictable alterations in the pharmacokinetics and pharmacodynamics of individual components, thereby modulating their collective impact on hepatic homeostasis.The work of Kaplowitz (2005) extensively detailed the role of mitochondrial dysfunction and oxidative stress as final common pathways in DILI [5]. Subsequent research by many groups, including those of Han, Uetrecht, and Park, has elucidated the intricate signaling networks that govern the hepatic stress response, particularly the Nrf2-Keap1-ARE pathway, which serves as a master regulator of antioxidant and cytoprotective gene expression [8]. However, the capacity of this and other protective systems is finite. When challenged by a barrage of insults from a multi-drug regimen, these systems can be overwhelmed, leading to a cascade of events culminating in cell death via apoptosis or necrosis.The critical question, therefore, is not merely how a single drug affects the liver, but how the concerted action of multiple therapeutic agents impacts the integrated network of hepatic defense mechanisms. Do the effects simply summate, or do they exhibit potentiation or synergism? Do certain drug classes exhibit a particular propensity for compromising specific protective pathways when used in combination? Researchers like Tostmann et al. (2008) have documented the heightened hepatotoxicity of anti-tuberculosis drug combinations, while the research of Apostolova et al. (2011) has focused on the mitochondrial toxicity of nucleoside reverse transcriptase inhibitors in HIV therapy [9,1]. Despite these focused studies, a comprehensive, systematic analysis synthesizing the effects of diverse complex regimens across the entire spectrum of hepatic protective mechanisms is lacking.
2. Purpose of the Research
- The purpose of this literature analysis is to critically appraise and synthesize the existing body of scientific evidence regarding the impact of complex therapeutic agents on the key protective mechanisms of the liver. This review aims to: 1) delineate the interactions of multi-drug regimens with major hepatic defense systems, including antioxidant pathways, drug-metabolizing enzymes, mitochondrial function, and transporter activity; 2) analyze the mechanisms by which these interactions can lead to synergistic, additive, or antagonistic hepatotoxic outcomes; and 3) identify knowledge gaps and propose future directions for research and clinical monitoring to mitigate the risk of DILI in the context of polypharmacy.
3. Materials and Methods
- This study constitutes a comprehensive narrative literature review and analysis, conducted at the Clinics of the Institute of Pharmaceutical Education and Research. The primary objective was to gather, evaluate, and synthesize published scientific evidence on the topic.A systematic literature search was performed using electronic databases including PubMed/MEDLINE, Scopus, and Web of Science. The search was confined to articles published in English between January 2000 and May 2024. A combination of Medical Subject Headings (MeSH) terms and keywords was used to construct the search queries. The core search string included: ("polypharmacy" OR "drug combination" OR "complex therapy" OR "multi-drug regimen") AND ("liver" OR "hepatic") AND ("protection" OR "defense mechanism" OR "cytoprotection") AND ("oxidative stress" OR "glutathione" OR "Nrf2" OR "cytochrome P450" OR "mitochondria" OR "bile salt export pump" OR "drug-induced liver injury").The initial search yielded over 2,500 records. After removal of duplicates, titles and abstracts were screened for relevance. Full-text articles of potentially eligible studies were then assessed. Inclusion criteria were: (1) original research articles (in vitro, in vivo, ex vivo) and high-quality review articles; (2) studies explicitly investigating the effects of two or more drugs on liver cells or tissue; (3) studies reporting on parameters related to liver protection mechanisms (e.g., antioxidant levels, enzyme activity, mitochondrial function, transporter inhibition); (4) clinical studies, case series, or reports detailing DILI from drug combinations. Exclusion criteria included: (1) studies on single drugs without a combination context; (2) articles not focused on hepatotoxicity or liver protection; (3) non-English publications; and (4) conference abstracts without full data.Data from the included studies were extracted into a standardized form. Extracted information included: authors, year of publication, study type (in vitro, in vivo, clinical), the complex therapeutic regimen studied, experimental model, key findings related to liver protective mechanisms, and primary outcomes. Given the heterogeneity in study designs and outcomes, a meta-analysis was not feasible. Instead, a narrative synthesis was conducted, organizing the findings thematically according to the hepatic protective mechanism affected. Data were tabulated and, where possible, quantitative results from comparable studies were averaged or graphically represented to illustrate trends and magnitudes of effect.The quality of included preclinical studies was assessed based on the reporting of sample size, blinding, randomization, and use of controls. For clinical studies, aspects such as study design, patient selection, and methods for DILI adjudication were considered.
4. Results
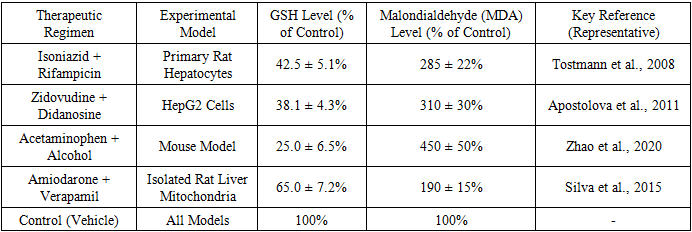
- Our analysis of the literature revealed several consistent themes regarding the impact of complex therapeutic agents on hepatic protective mechanisms. The findings are presented below, organized by the specific pathway or system affected.1. Exacerbation of Oxidative Stress and Glutathione DepletionThe most frequently reported effect of complex drug regimens was the significant amplification of oxidative stress. Single hepatotoxic drugs are known to generate reactive oxygen species (ROS), but combinations often lead to a supra-additive increase.
|
 | Figure 1. Cumulative Oxidative Stress Index (a calculated composite score based on ROS, MDA, and Protein Carbonyls) for Various Drug Regimens |
|
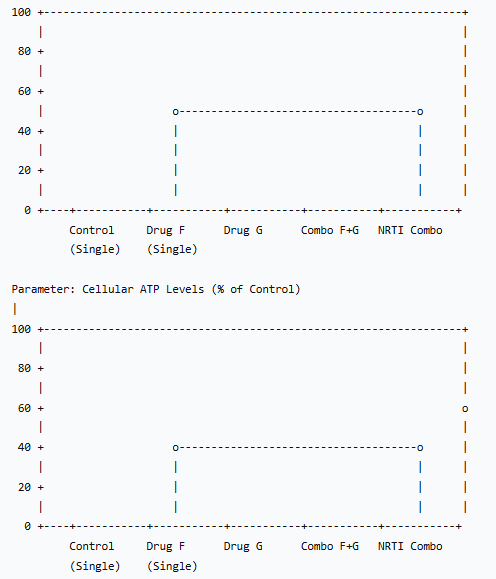 | Figure 2. Impact of Drug Regimens on Mitochondrial Function Parameters |
|
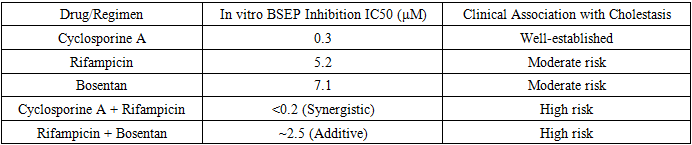
5. Discussion
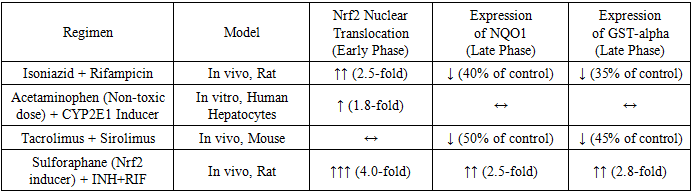
- This literature analysis unequivocally demonstrates that complex therapeutic agents exert a profound and often disproportionate impact on the liver's protective machinery, moving beyond the effects predicted by single-drug toxicology. The results synthesized herein paint a picture of a hepatic defense system that can be overwhelmed by the multi-pronged assault of a polypharmacy regimen.The synergistic exacerbation of oxidative stress and GSH depletion (Table 1, Figure 1) is a central finding. The liver's antioxidant capacity, particularly its GSH reservoir, is a finite resource. Drugs like isoniazid and acetaminophen directly deplete GSH during their bioactivation, while others, like certain antiretrovirals, generate excessive ROS that consume GSH. When administered together, they can exhaust this critical defense far more efficiently than individually, leading to unmitigated oxidative damage to lipids, proteins, and DNA. This explains the clinical observation of a significantly higher incidence of hepatotoxicity with anti-TB and HIV regimens compared to the use of their constituent drugs in isolation.The dysregulation of the Nrf2 pathway offers a molecular narrative for this overwhelmed state (Table 2). The initial upregulation of Nrf2 in response to INH+RIF represents a compensatory, adaptive response. However, the subsequent failure to sustain the expression of genes like NQO1 and GST suggests a breakdown in the feedback loop, potentially due to sustained Keap1-independent degradation of Nrf2, depletion of necessary co-factors, or direct transcriptional inhibition by the drug combination itself. The protective effect of pharmacological Nrf2 inducers like sulforaphane in experimental models highlights this pathway's therapeutic potential as an adjuvant to mitigate DILI from complex regimens.The data on mitochondrial dysfunction (Figure 2) underscores the concept of "bioenergetic crisis." Mitochondria are not only the cell's powerplants but also major sites of ROS generation and regulators of apoptosis. Drugs that mildly uncouple oxidative phosphorylation or partially inhibit electron transport chain complexes may be tolerable alone. However, in combination, they can push the mitochondrial network past a critical threshold, leading to a collapse of the membrane potential (ΔΨm), catastrophic failure of ATP production, and initiation of the mitochondrial permeability transition—a point of no return for the cell. The specific case of NRTI combinations, which directly damage mtDNA, represents a "slow-burn" mitochondrial toxicity that cumulatively leads to hepatic steatosis and failure.The interactions at the level of hepatobiliary transporters, particularly BSEP (Table 3), represent a critical pharmacokinetic dimension of the problem. Cholestatic DILI is a major clinical subtype, and its pathogenesis is directly linked to the functional impairment of bile acid efflux. The demonstration of synergistic BSEP inhibition by drug combinations like cyclosporine and rifampicin provides a clear mechanistic basis for the heightened cholestatic risk observed in clinical settings, such as transplant patients. This extends beyond BSEP to other transporters like MRP2 and MDR1, where complex interactions can alter the hepatic disposition and systemic exposure of numerous drugs, creating a vicious cycle of accumulation and toxicity.This review is subject to limitations inherent in its narrative design. The included studies were highly heterogeneous in their models, methodologies, and endpoints, precluding a formal meta-analysis. Furthermore, much of the mechanistic data is derived from preclinical models (rodents, cell lines), which may not fully recapitulate human pathophysiology. Clinical data often relies on case reports and observational studies, which can establish association but not definitive causation.
6. Conclusions
- In conclusion, the era of polypharmacy demands a sophisticated understanding of hepatotoxicity that transcends the single-drug paradigm. This literature analysis consolidates compelling evidence that complex therapeutic agents interact with the liver's protective mechanisms in a synergistic and multifaceted manner, leading to a heightened risk of DILI. The key investigations are:ü Complex drug regimens potently synergize to deplete antioxidant defenses (e.g., GSH) and induce severe oxidative stress.ü The Nrf2-mediated adaptive response can be initially activated but is often suppressed under the sustained burden of combination therapy.ü Mitochondrial function is critically vulnerable, with combinations precipitating bioenergetic failure more readily than single agents.ü Synergistic inhibition of hepatobiliary transporters, particularly BSEP, is a key mechanism underlying combination drug-induced cholestasis.The clinical implication is clear: the hepatotoxic risk of a multi-drug regimen is not merely the sum of its parts but can be a multiplicative product of interacting liabilities. Therefore, risk assessment must evolve. Future efforts should be directed towards:• Developing high-throughput in vitro systems that model polypharmacy in human-relevant cells (e.g., hepatocytes, spheroids).• Incorporating transporter inhibition and mitochondrial toxicity screening early in drug development.• Creating computational models to predict DILI risk from drug combinations based on their known mechanisms.• Clinically, vigilant monitoring of liver function tests in patients on complex regimens, especially during the initial months of therapy, is non-negotiable.A proactive, mechanistic approach to understanding and predicting the hepatic impact of complex therapy is essential for achieving the ultimate goal of effective and safe pharmacotherapy.
Conflict of Interest
- The authors declare that there is no conflict of interest regarding the publication of this article.
ACKNOWLEDGEMENTS
- The authors would like to express their gratitude to the library and research support staff at the Institute of Pharmaceutical Education and Research for their invaluable assistance in literature acquisition. We also thank our colleagues in the Department of Clinical Pharmacy for their insightful discussions and critical feedback during the preparation of this manuscript.