Lola T. Daminova, Asal S. Rashidova, Sitorakhon U. Muminova
Tashkent State Medical University, Tashkent, Uzbekistan
Correspondence to: Sitorakhon U. Muminova, Tashkent State Medical University, Tashkent, Uzbekistan.
| Email: |  |
Copyright © 2025 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Abstract
Glomerulonephritis (GN) comprises a group of kidney diseases characterized by immune-mediated glomerular injury leading to hematuria, proteinuria, and often progressive renal impairment. Immunosuppressive therapy is the cornerstone of GN management, aimed at controlling the underlying immune process and preserving kidney function. This review summarizes current approaches to immunosuppressive treatment in GN, from high-dose corticosteroid regimens (including pulse therapy) to cytotoxic agents and emerging targeted therapies. We discuss the indications and rationale for aggressive induction therapy in severe forms of GN (such as rapidly progressive GN), as well as maintenance strategies and supportive treatments. Advances in therapy—including optimized dosing strategies, risk-based treatment selection, and novel agents like targeted immunomodulators—have improved outcomes in diseases like IgA nephropathy. However, treatment-related toxicity remains a significant concern, underscoring the need for safer therapies. By selecting appropriate immunosuppressive regimens early, tailored to disease etiology and patient factors, it is possible to slow the progression to end-stage renal disease. Ongoing clinical trials and guidelines point toward a future of more personalized and less harmful treatments for GN.
Keywords:
Glomerulonephritis, Immunosuppressive therapy, Cyclophosphamide, IgA nephropathy
Cite this paper: Lola T. Daminova, Asal S. Rashidova, Sitorakhon U. Muminova, Immunosuppressive Therapy in Glomerulonephritis: Current State and Perspectives, American Journal of Medicine and Medical Sciences, Vol. 15 No. 11, 2025, pp. 3993-3997. doi: 10.5923/j.ajmms.20251511.54.
1. Introduction
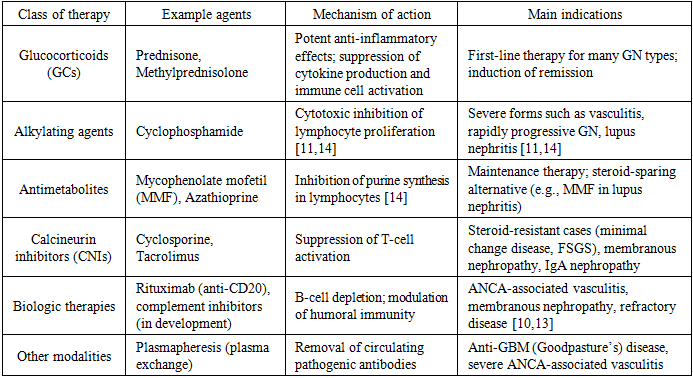
Glomerulonephritis represents a broad group of kidney disorders characterized by immune-driven inflammation and injury of the glomeruli. Clinically, it often manifests through hematuria, proteinuria, and azotemia. Around 85% of cases are classified as primary (idiopathic), while about 15% develop secondarily to systemic conditions such as systemic lupus erythematosus, vasculitis, chronic liver disease, or as a reaction to certain medications [1,3,4]. The clinical course of GN varies widely — from acute and potentially reversible forms to rapidly progressive (subacute) and chronic types that may culminate in chronic kidney disease. From a histopathological perspective, it encompasses distinct entities like minimal change disease, focal segmental glomerulosclerosis, membranous nephropathy, and IgA nephropathy, each with characteristic patterns of injury [3-5]. Epidemiologically, GN remains one of the major causes of end-stage renal disease worldwide, predominantly affecting men between their 30s and 40s, with a male-to-female ratio of roughly 2–3:1. Its incidence is estimated at 4–9 cases per 100,000 population annually, though actual rates may be higher due to underdiagnosis of mild or subclinical cases [1,6]. Globally, GN ranks third among the leading causes of chronic kidney disease, following diabetes and hypertension. Over the past three decades, the number of CKD cases associated with GN has risen by more than 80%, now affecting approximately 17–18 million people [2,7]. The annual incidence of primary GN is estimated at 0.2–2.5 cases per 100,000 people, with IgA nephropathy and membranous nephropathy being most common, especially in Asian populations [5,6]. This increase is largely attributed to improved diagnostic capabilities, population aging, and the growing prevalence of metabolic and infectious factors [7]. In Uzbekistan, national health data indicate that the prevalence of GN has increased from 8.3 to 12.7 cases per 10,000 population, mirroring the global trend [8]. Overall, GN continues to pose a significant medical and public health challenge that requires early detection and pathogenetically guided immunosuppressive therapy. Chronic GN (both primary and secondary) is responsible for approximately 10–15% of end-stage renal disease cases, reinforcing its role as the third most common cause of ESRD after diabetes and hypertension [1,2,7]. As a result, the underlying cause of GN often correlates with the severity of glomerular injury and the patient’s response to therapy.Immunopathogenesis is central in most GNs, making immunosuppressive therapy the cornerstone of treatment. Affected glomeruli typically show deposition of immune complexes or autoantibodies and complement activation, leading to inflammation [9], resulting in impaired renal function. Notably, some glomerular diseases (e.g. idiopathic membranous nephropathy) involve IgG4 subclass autoantibodies, indicating common mechanisms with other IgG4-related autoimmune diseases. For example, membranous GN shares pathogenic features with IgG4-mediated conditions such as pemphigus vulgaris and certain antibody-mediated encephalitides. Because of this immune basis, therapies that suppress or modulate the immune system are critical to prevent irreversible glomerular damage [11].Historically, glucocorticoids and cytotoxic agents have formed the backbone of GN therapy. High-dose corticosteroids (e.g. prednisone or methylprednisolone) can induce remission in many patients, especially those with minimal change disease or IgA nephropathy. Cytotoxic drugs like cyclophosphamide have been standard for aggressive forms such as lupus nephritis and ANCA-associated vasculitis. Over the past few decades, treatment strategies have evolved to maximize efficacy while minimizing toxicity. Notably, in 1976 Cathcart et al. introduced the concept of “pulse” corticosteroid therapy – very high doses of methylprednisolone given intravenously in short courses – reporting successful reversal of severe lupus nephritis with rapidly deteriorating renal function. This innovation laid the groundwork for treating life-threatening rheumatic diseases and rapidly progressive GN with intermittent high-dose immunosuppression. Today, immunosuppressive regimens in GN include a spectrum from conventional agents (corticosteroids, alkylating agents, antimetabolites, calcineurin inhibitors) to newer biologic therapies (e.g. rituximab) and targeted therapies (e.g. complement inhibitors), guided by disease type and severity [12]. In this review, we provide an overview of the current state of immunosuppressive therapy in glomerulonephritis. We have structured the discussion to cover the major therapeutic approaches, their evidence base, and recent developments. We also highlight the importance of early and appropriate therapy selection—tailored to the GN subtype and patient factors—in improving renal outcomes.The primary goal of immunosuppressive therapy in glomerulonephritis is to induce and maintain remission of active disease, thereby preserving renal function and preventing progression to chronic kidney failure. Because GN often results from an overactive or aberrant immune response, therapies that attenuate immune activity can halt the inflammatory damage in glomeruli. The choice of therapy and intensity depends on the specific GN diagnosis and severity of disease. For instance, mild forms of GN (e.g. early IgA nephropathy with low risk) might be managed with supportive care and possibly low-dose steroids, whereas severe forms (e.g. crescentic GN with rapid renal decline) require aggressive immunosuppression such as high-dose steroids plus cytotoxic drugs. It is crucial to balance efficacy with toxicity: high-intensity regimens are given for severe disease (induction phase) and then tapered or switched to safer maintenance regimens once remission is achieved [11,13].Immunosuppressants used in GN include glucocorticoids, cytotoxic alkylating agents, antimetabolites, calcineurin inhibitors, and biologic agents:Table 1. Major classes of immunosuppressive therapy in glomerulonephritis
 |
| |
|
Conventional corticosteroid therapy: High-dose corticosteroids are often the first-line immunosuppressive treatment in many glomerulonephritides. Prednisone (or an equivalent) is typically given at doses of 0.5–1 mg/kg/day (up to ~60 mg daily in adults) for several weeks, then tapered over months [14]. This approach can induce remission in diseases like idiopathic minimal change disease (where steroids induce remission in ~80% of adults) [15] and is a component of therapy in lupus nephritis, IgA nephropathy with significant proteinuria, and vasculitic GN. The anti-inflammatory and immunosuppressive effects of steroids are broad: they reduce leukocyte infiltration, cytokine production, and antibody formation. However, prolonged high-dose steroid use is associated with numerous adverse effects (Cushingoid features, hyperglycemia, hypertension, infections, osteoporosis, etc.). Therefore, clinicians aim to use the minimum effective dose and duration. Adjunct therapies often seek to spare or replace steroids due to these toxicities [15,16]. Pulse therapy refers to the intermittent administration of above therapeutic doses of drugs over short periods to maximize therapeutic impact while potentially reducing long-term side effects. In the context of GN, pulse corticosteroid therapy usually means intravenous methylprednisolone in very high doses given for a few days. The concept was pioneered in the 1970s: E. S. Cathcart et al. reported that ultra-high-dose IV methylprednisolone (e.g. 1 gram daily) for a brief course improved outcomes in patients with lupus nephritis and rapidly worsening renal function. Since then, steroid pulses have been widely adopted as an induction strategy for severe autoimmune glomerular diseases. Indications for pulse steroids include life-threatening or organ-threatening situations, such as rapidly progressive GN with crescent formation, severe ANCA-associated vasculitis, lupus nephritis with acute renal failure, or other systemic autoimmune diseases with critical manifestations. Pulse therapy is typically used in the induction phase to achieve rapid disease control (remission induction stage) before irreversible tissue damage occurs. It is often combined with other immunosuppressives.Clinical evidence supports pulse steroids in severe GN. For example, in ANCA-associated vasculitis with renal involvement, pulse IV methylprednisolone for 3 days is commonly given at presentation before starting cyclophosphamide or rituximab. In lupus nephritis, guidelines often recommend a pulse of methylprednisolone (500–1000 mg daily for 3 doses) at the start of induction therapy. Pulse steroids have also been used in other acute settings like severe COVID-19 pneumonia to curb hyperinflammation: a randomized trial reported that intravenous methylprednisolone pulses improved outcomes in hospitalized patients with severe COVID-19. Similarly, a comparative study in critical COVID-19 found that pulse-dose corticosteroids could be beneficial versus lower-dose regimens. This underscores that the pulse therapy concept—first developed in GN and rheumatology—has broad applicability in acute inflammatory conditions [17]. Even in pulse form, corticosteroids can cause significant side effects. Short-term pulses can lead to transient blood pressure elevation, hyperglycemia, mood changes, or sleep disturbances. With repeated pulses or prolonged high-dose use, patients may experience severe infections (due to immunosuppression), steroid-induced diabetes, psychosis, or avascular necrosis. Notably, high-dose steroids can induce Cushing’s syndrome signs (weight gain, moon facies, etc.) and hypothalamic-pituitary-adrenal axis suppression. Cases of steroid pulse therapy have reported rare complications such as arrhythmias and even sudden cardiac death (though causality is unclear). Thus, while pulses are invaluable for induction, they are usually given for short durations and always with careful monitoring. Maintenance therapy then transitions to safer doses or alternative agents. Cyclophosphamide (CYC) is a potent alkylating agent that has long been a cornerstone of induction therapy for severe glomerulonephritis, including pauci-immune crescentic GN (ANCA-associated vasculitis), severe lupus nephritis (Class III/IV), and other aggressive forms [14]. It acts by crosslinking DNA and inducing apoptosis of proliferating lymphocytes, resulting in strong immunosuppression [14]. CYC can be administered either orally (1.5–2 mg/kg/day for 3–6 months) or as monthly intravenous pulses (0.5–0.75 g/m²), with EUVAS trials showing comparable efficacy and lower cumulative toxicity for the pulse regimen [2,3]. In severe disease, pulse cyclophosphamide is often combined with high-dose corticosteroid pulses to rapidly induce remission, typically at a dose of 1 g IV every 4 weeks, with or without concomitant methylprednisolone 1 g IV [18]. Despite its efficacy, cyclophosphamide has significant adverse effects, including bone marrow suppression, infertility, hemorrhagic cystitis, increased infection risk, and long-term malignancy risk; therefore, it is generally used for 3–6 months for induction, followed by a safer maintenance agent such as azathioprine or mycophenolate [19]. Its use requires careful patient selection, close monitoring, and preventive measures to mitigate toxicity. Rituximab, an anti-CD20 monoclonal antibody, is an effective therapy for several glomerular diseases. In ANCA-associated vasculitis, RAVE and RITUXVAS trials showed it is at least as effective as cyclophosphamide and often preferred to avoid CYC toxicity. It is typically given as 375 mg/m² weekly for 4 doses or 1 g on days 1 and 15. Rituximab is also effective in idiopathic membranous nephropathy (targeting PLA2R autoantibodies) and is used in refractory lupus nephritis and some severe IgA nephropathy cases. Calcineurin inhibitors (CNIs) such as cyclosporine A and tacrolimus suppress T-cell activation and are effective in steroid-resistant or dependent MCD and FSGS, as well as membranous nephropathy and lupus nephritis [20]. Tacrolimus often has more predictable absorption and fewer cosmetic effects than cyclosporine. Their main limitation is nephrotoxicity, along with hypertension, tremors, hyperglycemia, and cosmetic side effects. Therefore, CNIs are used at the lowest effective dose and for limited duration with close monitoring [21]. IgA nephropathy (IgAN) is the most common primary glomerulonephritis worldwide, and its treatment strategy is rapidly evolving. Traditionally, therapy relied on supportive care with RAAS blockade and corticosteroids for high-risk cases, but steroid toxicity has limited their use. Current KDIGO 2021 guidelines emphasize risk stratification using clinical and histologic data to guide treatment intensity. New options include targeted budesonide, SGLT2 inhibitors (e.g., dapagliflozin), and biologic agents such as sibeprenlimab and telitacicept. The TESTING trial (steroids), MMF trials in Asia, and the Phase 3 sparsentan trial have shown encouraging results, signaling a shift toward more personalized and safer therapies for IgAN [5,9,14].Other GN subtypes: In membranous nephropathy, prolonged cyclophosphamide/steroid regimens are being replaced by rituximab, which achieves high remission rates and is now first-line for many patients. In minimal change disease and FSGS, steroids remain first-line, with CNIs or cyclophosphamide for relapses or resistant cases; ACTH gel is an option for refractory disease [10,13]. C3 glomerulopathies respond poorly to standard therapy, and complement inhibitors like pegcetacoplan are under study. In lupus nephritis, induction usually involves cyclophosphamide or MMF with steroids, and maintenance with MMF or azathioprine. Adding biologics such as belimumab (BLISS-LN) or voclosporin (AURORA) improves renal outcomes. Overall, therapy is increasingly personalized, balancing efficacy and toxicity according to disease type, severity, and patient factors [11]. Adverse Effects and Management Considerations: Immunosuppressive therapy in glomerulonephritis carries significant risks, particularly infections, drug-specific toxicities, and metabolic complications [11,14]. Patients on high-dose steroids or cytotoxic drugs require Pneumocystis prophylaxis and updated vaccinations prior to therapy, with close monitoring for opportunistic infections [14]. Routine safety monitoring includes blood counts, liver enzymes, blood glucose, and cancer surveillance in those previously exposed to cyclophosphamide. To minimize toxicity, modern protocols favor lower doses and shorter exposure—for example, the Euro-Lupus protocol uses [12,18] reduced cyclophosphamide doses, and ANCA vasculitis maintenance increasingly relies on rituximab or azathioprine instead of prolonged cyclophosphamide. Long-term steroids can lead to avascular necrosis, metabolic syndrome, and osteoporosis; bone protection with calcium, vitamin D, and bisphosphonates is recommended. Gonadoprotective measures may be considered for young patients on alkylating agents. Effective management requires multidisciplinary collaboration and adherence to KDIGO 2021 guidelines, which emphasize individualized risk reduction and integration of emerging therapies such as SGLT2 inhibitors and biologics [14,15].
2. Conclusions
Immunosuppressive therapy remains the cornerstone of glomerulonephritis management, with timely, individualized treatment improving renal outcomes and quality of life. Advances such as rituximab for ANCA-associated GN and membranous nephropathy, along with emerging targeted therapies including complement and cytokine inhibitors, are transforming care. There is a growing shift toward personalized approaches guided by risk prediction tools and biomarkers. Future therapies aim to provide durable remission with fewer adverse effects, reflecting rapid progress in precision immunosuppression.
References
| [1] | Kazi AM, Hashmi MF. Glomerulonephritis. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560644/. |
| [2] | Global Burden of Disease Collaborative Network. Global Burden of Kidney Disease Study 1990–2019. Seattle, WA: Institute for Health Metrics and Evaluation (IHME); 2020. Available from: http://ghdx.healthdata.org/gbd-results-tool. |
| [3] | Floege J, Amann K. Primary glomerulonephritides. Lancet. 2016; 387(10032): 2036–48. doi: 10.1016/S0140-6736(16)00272-5. |
| [4] | Couser WG. Glomerulonephritis. Lancet. 1999; 353(9163): 1509–15. doi: 10.1016/S0140-6736(98)08475-6. |
| [5] | Lv J, Zhang H. IgA nephropathy in China: progress and challenges. Am J Kidney Dis. 2016; 68(5): S50–S56. doi: 10.1053/j.ajkd.2016.05.021. |
| [6] | Glassock RJ. The epidemiology of glomerulonephritis. Nephron Clin Pract. 2010; 116(4): c305–c310. doi: 10.1159/000319452. |
| [7] | Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, et al. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet. 2013; 382(9889): 339–52. doi: 10.1016/S0140-6736(13)60595-4. |
| [8] | Министерство здравоохранения Республики Узбекистан. Национальный отчёт по состоянию нефрологических заболеваний. Ташкент; 2022. |
| [9] | Anders HJ, Palmen J, Vielhauer V. Glomerulonephritis: immunopathogenesis and new classification. Clin J Am Soc Nephrol. 2023; 18(2): 161–171. doi: 10.2215/CJN.0000000000000000. |
| [10] | Ronco P, Beck L, Debiec H, Fervenza FC, Hou FF, Jha V, et al. Membranous nephropathy: pathogenesis and treatment. Nat Rev Dis Primers. 2021; 7(1): 69. doi: 10.1038/s41572-021-00303-z. |
| [11] | Kant S, Jha V, Abbas H. Advances in understanding of pathogenesis and immune-mediated kidney disease. Am J Kidney Dis. 2022; 79(5): 736–748. doi: 10.1053/j.ajkd.2021.11.014. |
| [12] | Puéchal X, Iudici M, Perrodeau E, et al. Rituximab vs Cyclophosphamide Induction Therapy for Patients With Granulomatosis With Polyangiitis. JAMA Netw Open. 2022; 5(11): e2243799. doi: 10.1001/jamanetworkopen.2022.43799 |
| [13] | Trujillo H, et al. Ten tips on immunosuppression in primary membranous nephropathy. Clin Kidney J. 2024. |
| [14] | Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021; 100(4S): S1–S276. |
| [15] | Ponticelli C, Glassock RJ. Glucocorticoids in primary glomerular diseases: current status and future perspectives. J Nephrol. 2020; 33(6): 1161–1173. |
| [16] | Faulhaber-Walter R, et al. Glucocorticoids in glomerulonephritis: mechanisms of action and clinical use. Nat Rev Nephrol. 2024; 20(2): 78–91. |
| [17] | Rovin BH, Adler SG, Barratt J, et al. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases: lupus nephritis. Kidney Int. 2021; 100(4S): S1–S276. |
| [18] | Terrier B, et al. Rituximab versus conventional therapy for remission in ANCA-associated vasculitis (REOVAS). Ann Intern Med. 2025. doi: 10.7326/ANNALS-24-03947. |
| [19] | Ysermans R, et al. Adding low-dose cyclophosphamide to rituximab for remission induction in ANCA vasculitis. BMC Nephrol. 2022; 23(1): 416. |
| [20] | Lee HJ, et al. Tacrolimus and cyclosporine: comparison of efficacy, mechanism and toxicity. PMC. 2023. |
| [21] | Karolin A, Escher G, Rudloff S, Sidler D. Nephrotoxicity of Calcineurin Inhibitors in Kidney Epithelial Cells is Independent of NFAT Signaling. Frontiers in Pharmacology. 2022. doi: 10.3389/fphar.2021.789080. |

 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML