Xusanova Diyora Ziyadullaevna, Ibragimova Sapura Zaxidovna, Rizaeva Feruza Abdulxamitovna, Babakhanova Nargiza Nusratullayevna, Aripova Nazokat Bakhodirovna, Erimbetova Indira Oralbayevna, Kleevleva Albina Rustamovna
Scientific Practical Center of Pediatric Oncology, Hematology and Immunology, Tashkent, Uzbekistan
Copyright © 2025 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Abstract
Hereditary thrombocytopenias such as Bernard-Soulier syndrome are rare but are often misdiagnosed as ITP due to similar clinical presentation. The article describes 17 patients with chronic recurrent ITP who underwent NGS testing due to poor response to therapy. Of the 17 patients, 5 were found to have various forms of hereditary thrombocytopenia, while the remaining 11 cases were found to have pathological and conditionally pathological genes, the clinical presentation of which was combined with thrombocytopenia and hemorrhagic syndrome, in many cases resistant to platelet concentrate transfusion. Also presented are cases from the practice of patients who received treatment for chronic ITP with no effect or temporary effect, which was accompanied by severe hemorrhagic syndrome.
Keywords:
Idiopathic thrombocytopenic purpura (ITP), Children, Hereditary thrombocytopenias, Next generation sequencing (NGS)
Cite this paper: Xusanova Diyora Ziyadullaevna, Ibragimova Sapura Zaxidovna, Rizaeva Feruza Abdulxamitovna, Babakhanova Nargiza Nusratullayevna, Aripova Nazokat Bakhodirovna, Erimbetova Indira Oralbayevna, Kleevleva Albina Rustamovna, Results of Next-Generation Sequencing Studies in Children with Chronic ITP (Single Center Experience), American Journal of Medicine and Medical Sciences, Vol. 15 No. 10, 2025, pp. 3279-3282. doi: 10.5923/j.ajmms.20251510.01.
1. Introduction
The most common cause of isolated thrombocytopenia in children is idiopathic thrombocytopenic purpura (ITP). Hereditary thrombocytopenias such as Bernard-Soulier syndrome are rare but are often misdiagnosed as ITP due to their similar clinical presentation. Hereditary macrothrombocytopenia is a rare disorder that is often misdiagnosed as idiopathic immune thrombocytopenia (ITP). Automated blood cell counters in routine clinical practice commonly miss giant platelets and underestimate the mean platelet volume (MPV). Misdiagnoses may expose patients to unnecessary immunosuppressive treatment [1,2]. Therefore, genetic testing in the form of whole genome or whole exome sequencing can help to accurately diagnose and determine the correct treatment strategy. Hereditary thrombocytopenia (HIT) is an example of a common misdiagnosis; up to 40% of patients with HIT are initially misdiagnosed as chronic ITP [3].Goal. Assess the significance and role of using next generation sequencing (NGS) research in chronic forms of ITP in children.
2. Materials and Methods
Peripheral blood, case history materials of 17 patients with chronic ITP. Average age 5.6 years, girls - 7, boys - 10. The following were performed: peripheral blood, bone marrow - hemogram, myelogram, whole exome sequencing of peripheral blood DNA, clinical examination, family history, if necessary, additional radiological and biochemical studies [4,5].The laboratory of the study was carried out in the optimum conditions recommended on the kit, and the raw data obtained were made ready for analysis by going through a series of processes. Current versions of different databases were used for interpretation of variants during the analyses. Mutations with a minor allele frequency greater than 5% were note valuated. Detected variants were classified according to ACMG (American College of Medical Genetics and Genomics) criteria published in 2015. As a result of the analyzes, pathogenic, possibly pathogenic and clinically unknown variants associated with the patient's clinic were reported. In addition, even if it is not related to the patient's clinic, pathogenic and possibly pathogenic variants detected in the genes recommended to be reported by ACMG were added to the report [6].The patients were searched for pathogenic variants of the nucleotide sequence associated with hereditary thrombocytopenia and other diseases with similar phenotypic manifestations.
3. Discussion
NGS study was conducted in 17 patients with chronic recurrent ITP, due to poor response to therapy. The results are presented in Table 1. 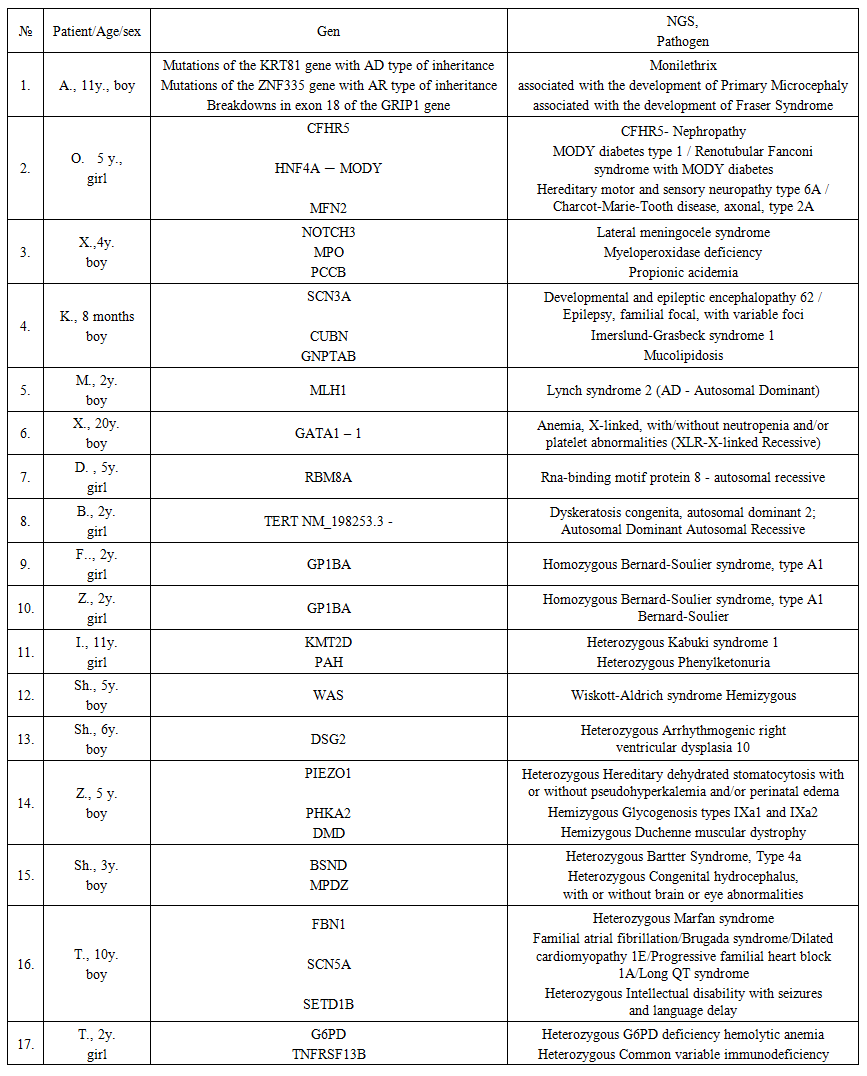 | Table 1. Characteristics of the examined patients and interpretation of the identified genes |
Of the 17 patients, 5 were found to have various forms of hereditary thrombocytopenia, such as Homozygous Bernard-Soulier syndrome – 2 patients, Wiskott-Aldrich syndrome Hemizygous - 1, Heterozygous Kabuki syndrome – 1, Anemia, X-linked, with/without neutropenia and/or platelet abnormalities (XLR-X-linked Recessive) – 1.While the remaining 11 cases were found to have pathological and conditionally pathological genes, the clinical presentation of which was combined with thrombocytopenia and hemorrhagic syndrome, in many cases resistant to platelet concentrate transfusion. Below are the cases from practice, these are patients who received treatment for chronic ITP with no outcome for treatment, accompanied by severe hemorrhagic syndrome, which required, in some cases, platelet concentrate transfusions.Clinical cases.1. Girls, twins, 2 years old, were hospitalized in the 1st oncohematology department of the Scientific and Practical Medical Center for pediatric oncology, hematology and immunology with the diagnosis: ITP, chronic recurrent form, hemorrhagic syndrome. Complaints upon admission about the appearance of bruises and nosebleeds.Anamnesis morbi: The mother told, they have been sick since 4 months of age. The disease began acutely, a few hours after vaccination. Multiple petechiae and small bruises appeared on the skin, nosebleeds. Received hemostatic therapy and transfusions of fresh frozen plasma in the hospital at the place of residence. In the blood test from 08/11/2023, platelets were 18.0 thousand. In the myelogram from 21.08.2023 blasts - 4.0%, lymphocytes - 10.8%, bone marrow punctate is cellular, there are many megakaryocytes, 24 were examined, of which: with platelet pinching - 3, platelet-containing - 2, platelet-free - 6, naked - 13.They received intravenous immunoglobulin in a course dose of 10.0 grams, prednisolone 10 mg orally from 08/14/20203, ceftriaxone, symptomatic treatment.In the blood test from 08/23/2023: hemoglobin - 111.0 g / l, erythrocytes - 4.02 million, platelets - 14.0 thousand, leukocytes - 8.97 thousand, neutrophils - 3.91 thousand, ESR - 2 mm / h.After discharge, they were repeatedly treated in the hospital at the place of residence, frequent nosebleeds were noted. Repeated hospitalization at the Hematology Center from 05/27/2024 to 06/15/20204. with a diagnosis: Idiopathic thrombocytopenic purpura, chronic relapsing form, hemorrhagic syndrome. Severe posthemorrhagic anemia. Sepsis. Bilateral bronchopneumonia. Delayed speech and physical development. They received platelet concentrate No. 2, erythrocyte mass No. 1, octagam in a course dose of 15.0 grams for 3 days, prednisolone 15 mg for 3 days, methylprednisolone 32 mg orally, revoleid, meropenem. In the coagulogram from 10/2/2024, APTT-32 sec., PTI-105%, fibrinogen-2.91 g/l. Indirect Coombs test from 10/2/2024 positive 1:8, transfusions - platelet concentrate No. 1. The condition has improved, the bleeding has stopped.Blood test for NGS taken on 06/01/2024. The patients were searched for pathogenic variants of the nucleotide sequence associated with idiopathic thrombocytopenic purpura, Fanconi aplastic anemia, and other hereditary diseases with similar phenotypic manifestations. A previously undescribed variant of the nucleotide sequence in exon 3 of the GP1BA gene in the homozygous state was identified, leading to premature termination of protein translation. The GP1BA gene encodes the alpha subunit of Glycoprotein Ib, a glycoprotein of the platelet surface membrane that functions as a receptor for the von Willebrand factor [1-2]. Mutations in the GP1BA gene with the AR type of inheritance are associated with the development of Bernard-Soulier syndrome, type A1 [5,6]. Based on the NGS results from 08/08/2024, the diagnosis was: Bernard-Soulier syndrome type A1. 2. The patient, a girl, aged 10, was undergoing inpatient treatment at the Scientific and Practical Center for Pediatric Oncology, Hematology and Immunology with the diagnosis: Immune thrombocytopenic purpura, chronic relapsing form. D69.3. Generalized hemorrhagic syndrome. Concomitant: Congenital heart defect, atrial septal defect. Congenital dislocation of the hip joints.Complaints upon admission: weakness, lethargy, decreased appetite, pale skin, the appearance of multiple hemorrhages and bruises on the body and bruises at the injection sites, profuse nosebleeds. Anamnesis morbi: the patient was diagnosed with delayed speech development by a neurologist since 2017. The operation was performed in 2016 in India with the diagnosis: Congenital heart defect, atrial septal defect. The operation was performed with the diagnosis: Congenital dislocation of the hip joints. For the last 2 days, profuse gum bleeding was noted at the site of the extracted tooth, the patient was referred for further examination and treatment to the outpatient clinic of the Center and with the diagnosis: ITP? To exclude a systemic blood disease, she was hospitalized in the department. Then, from 09/04/2024 to 09/20/2024, she received inpatient treatment with the diagnosis: Idiopathic thrombocytopenic purpura, acute course. Received antibiotic therapy; antifungal therapy; hemostatic therapy; Octagam 1g-20 ml intravenous drip No. 16 bottles of 4 bottles No. 4 days; Laboratory data: blood test from 11.03.25. gem.-132.0 g / l; erythr.-4.92; thrombus-5.0 thousand; leukocytes-5.27; granulocytes-3.28; p / y-2; s / y-61; lymph.-30; mon.-6; eos.-1; ESR-6 mm / h; Blood coagulation profile from 12.03.25. Platelet aggregation with ADP-3%; APTT-55 sec.; PTI-100%; FP-2.5; Ultrasound of abdominal organs from 03/14/25. Conclusion: Diffuse changes in the liver parenchyma. Diffuse and focal changes in the spleen parenchyma.NGS study was performed. Conclusion from 06/27/2024 A previously described variant of the nucleotide sequence in exon 39 of the KMT2D gene was identified in a heterozygous state, leading to premature translation termination. The KMT2D gene encodes histone methyltransferase, which methylates histone H3. The encoded protein is part of a large protein complex called ASCOM, which has been shown to be a transcriptional regulator of the beta-globin and estrogen receptor genes [7]. Mutations in the KMT2D gene with the AD inheritance pattern are associated with the development of Kabuki syndrome. The detected variant was previously described in patients with Kabuki syndrome and was registered in the gnomAD control sample with a frequency of 0.00006199% (on 1 chromosome out of 1,613,048). Based on the totality of information, the detected variant of the nucleotide sequence should be regarded as a pathogenic variant. The patient is also a carrier of Phenylketonuria - A previously undescribed variant of the nucleotide sequence in exon 7 of the PAH gene was detected in a heterozygous state, leading to a missense substitution of the amino acid in codon 243. The PAH gene encodes Phenylalanine hydroxylase, which catalyzes the hydroxylation of phenylalanine to tyrosine, the rate-limiting step in phenylalanine catabolism. Mutations in the PAH gene with the AR type of inheritance are associated with the development of Phenylketonuria and was not registered in the gnomAD control sample. Then she received Hormone therapy - prednisolone - 5 mg at the rate of 2 mg / kg for a total of 45 mg orally according to the scheme (short courses); IVIG Bioven 2.5 g - 50 mg intravenously drip 9 bottles 3 + 3 + 3 bottle according to the scheme, Eltrombopag tab. 25 mg orally. As a result of the therapy, the patient’s condition improved and the hemorrhagic syndrome was relieved.Kabuki syndrome (KS) is a clinically recognisable syndrome in which 70% of patients have a pathogenic variant in KMT2D or KDM6A. Understanding the function of these genes opens the door to targeted therapies. Kabuki syndrome (KS) is a dominantly inherited disorder mainly due to de novo pathogenic variation in KMT2D or KDM6A genes. We conclude that KMT2D sequencing followed by array CGH is a diagnostic strategy with the highest diagnostic yield. The field of dysmorphology has been changed by the use Artificial Intelligence (AI) and the development of Next Generation Phenotyping (NGP) [8,9]. Based on the NGS results from 06/27/2024, the diagnosis was: Kabuki syndrome (KS).
4. Conclusions
All 17 patients were initially diagnosed with immune thrombocytopenia, and received therapy with intravenous immunoglobulins and steroids. In these patients, the clinic had a pronounced hemorrhagic syndrome, hereditary burden, and early onset of the disease, which, according to international recommendations, is an indication for NGS testing. This contributed to the timely diagnosis and avoided unnecessary toxicity and the use of third-line drugs such as rituximab, cyclosporine, and mycofolenate mofetil, which are used to treat chronic ITP. Establishing an accurate diagnosis also made it possible to identify patients who need further allogeneic bone marrow transplantation. For patients for whom there is no curative treatment due to the hereditary nature of the disease, it improved the quality of life and determined the prognosis.
References
| [1] | Kottayam R, Rozenberg G, Brighton T, Cohn RJ. Isolated thrombocytopenia in children: thinking beyond idiopathic thrombocytopenic purpura and leukaemia. J Paediatr Child Health. 2007 Dec; 43(12): 848-50. doi: 10.1111/j.1440-1754.2007.01240.x. PMID: 18036021. |
| [2] | Gohda F, Uchiumi H, Handa H, Matsushima T, Tsukamoto N, Morita K, Amagai H, Murakami M, Murakami H, Nojima Y, Karasawa M. Identification of inherited macrothrombocytopenias based on mean platelet volume among patients diagnosed with idiopathic thrombocytopenia. Thromb Res. 2007; 119(6): 741-6. doi: 10.1016/j.thromres.2006.06.011. Epub 2006 Aug 17. PMID: 16916536. |
| [3] | Doubek M, Smejkal P, Dostálová V, Trnavská I, Buliková A, Brychtová Y, Mayer J. Dĕdicné trombocytopenie. Diferenciální diagnostika jednoho prípadu [Hereditary thrombocytopenia. Differential diagnosis of a case]. Cas Lek Cesk. 2003; 142(11): 683-6. Czech. PMID: 14689830. |
| [4] | Balduini CL, Melazzini F, Pecci A. Inherited thrombocytopenias-recent advances in clinical and molecular aspects. Platelets. 2017; 28(1): 3-13. doi:10.3109/09537104.2016.1171835. |
| [5] | Sumitha E, Jayandharan GR, David S, et al. Molecular basis of Bernard– Soulier syndrome in 27 patients from India. J Thromb Haemost. 2011; 9(8): 1590-1598. doi:10.1111/j.1538-7836.2011.04417.x. |
| [6] | Favier R, Raslova H. Progress in understanding the diagnosis and molecular genetics of macrothrombocytopenias. Br J Haematol. 2015; 170(5): 626-639. PubMed|https://doi.org/10.1111/bjh.13478|Google Scholar. |
| [7] | Adam MP, Banka S, Bjornsson HT, Bodamer O, Chudley AE, Harris J, Kawame H, Lanpher BC, Lindsley AW, Merla G, Miyake N, Okamoto N, Stumpel CT, Niikawa N; Kabuki Syndrome Medical Advisory Board. Kabuki syndrome: international consensus diagnostic criteria. J Med Genet. 2019 Feb; 56(2): 89-95. doi: 10.1136/jmedgenet-2018-105625. Epub 2018 Dec 4. PMID: 30514738. |
| [8] | Molecular genetic analysis in 14 Czech Kabuki syndrome patients is confirming the utility of phenotypic scoring. Paděrová J, Holubová A, Simandlová M, Puchmajerová A, Vlčková M, Malíková M, Pourová R, Vejvalková S, Havlovicová M, Šenkeříková M, Ptáková N, Drábová J, Geryk J, Maver A, Křepelová A, Macek M Jr. Clin Genet. 2016 Sep; 90(3): 230-7. doi: 10.1111/cge.12754. Epub 2016 Mar 8. PMID: 26841933. |
| [9] | Next generation phenotyping for diagnosis and phenotype-genotype correlations in Kabuki syndrome. Hennocq Q, Willems M, Amiel J, Arpin S, Attie-Bitach T, Bongibault T, Bouygues T, Cormier-Daire V, Corre P, Dieterich K, Douillet M, Feydy J, Galliani E, Giuliano F, Lyonnet S, Picard A, Porntaveetus T, Rio M, Rouxel F, Shotelersuk V, Toutain A, Yauy K, Geneviève D, Khonsari RH, Garcelon N. Sci Rep. 2024 Jan 28; 14(1): 2330. doi: 10.1038/s41598-024-52691-3. |

 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML