-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2025; 15(5): 1325-1330
doi:10.5923/j.ajmms.20251505.02
Received: Feb. 16, 2025; Accepted: Mar. 12, 2025; Published: May 8, 2025

Case of Acute Lymphoblastic Leukemia with del(13)(q14)
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLYulianna Yurevna Assesorova 1, Timur Raufovich Alimov 1, Irodakhon Tulanovna Kodirova 2, Kodirzhon Tukhtaboevich Boboev 3
1Molecular Genetics, Cytogenetics & FISH Laboratory, Republican Specialized Scientific-Practical Medical Center of Hematology (RSSPMCH) MoH RUz, Tashkent, Uzbekistan
2Department of Hematology (Clinic), Republican Specialized Scientific-Practical Medical Center of Hematology (RSSPMCH) MoH RUz, Tashkent, Uzbekistan
3Medical Genetics Laboratory, Republican Specialized Scientific-Practical Medical Center of Hematology (RSSPMCH) MoH RUz, Tashkent, Uzbekistan
Correspondence to: Timur Raufovich Alimov , Molecular Genetics, Cytogenetics & FISH Laboratory, Republican Specialized Scientific-Practical Medical Center of Hematology (RSSPMCH) MoH RUz, Tashkent, Uzbekistan.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Introduction. Acute B-lymphoblastic leukemia (B-ALL) is classified into subtypes based on certain recurrent chromosomal abnormalities. However, in some cases, atypical chromosomal rearrangements detected in В-ALL, as well as the features of the clinical picture of the disease, make diagnosis difficult. The purpose of this report is to describe the clinical observation of a patient with B-ALL, which was characterized by clinical features, hyperdiploid karyotype and del(13)(q14). Clinical observation. The article describes a clinical case of a 30-year-old man with Common (BII) ALL. The peculiarity of this case was the development of deep anemia and thrombocytopenia in the onset of the disease, as well as the formation of lytic foci in the bones. High blastosis, immunophenotype CD10+CD19+CD79a+TdT+CD34+ typical for B-ALL, and also hyperdiploid karyotype were established in the terminal period of the disease. Bone marrow examination revealed del(13)(q14). Conclusion. The described case shows that the spectrum of genetic changes in B-ALL with the typical immunophenotype CD10+CD19+CD79a+TdT+CD34+ may include not only recurrent genetic abnormalities that determine the subtypes of the disease, but also other chromosomal rearrangements, the diagnostic and prognostic significance of which will be established in the future in process of accumulation of data.
Keywords: Acute lymphoblastic leukemia, Osteolytic bone damage, Deletion (13)(q14), Hyperdiploidy
Cite this paper: Yulianna Yurevna Assesorova , Timur Raufovich Alimov , Irodakhon Tulanovna Kodirova , Kodirzhon Tukhtaboevich Boboev , Case of Acute Lymphoblastic Leukemia with del(13)(q14), American Journal of Medicine and Medical Sciences, Vol. 15 No. 5, 2025, pp. 1325-1330. doi: 10.5923/j.ajmms.20251505.02.
Article Outline
1. Introduction
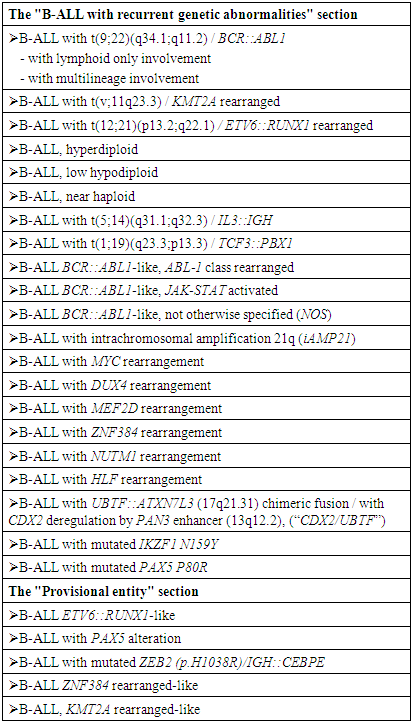
- Acute lymphoblastic leukemia / acute lymphoblastic lymphomas are represented by a heterogeneous group of malignant clonal diseases of the blood system originating from hematopoiesis progenitor cells of predominantly lymphoid differentiation. Most often, lymphoblastic neoplasm proceeds as a generalized leukemic process with primary damage to the bone marrow, displacement of normal hematopoiesis and involvement of various organs and body systems in the process. But it can also be manifested by localized infiltration of tissues with limited involvement of the bone marrow [1,2]. According to the updated International Consensus Classification (ICC) [3] acute-lymphoblastic leukemia (B-ALL) includes nosological variants that are stratified based on data of cytogenetic, molecular genetic studies and immunophenotyping (Table 1). These subtypes are associated with different disease outcomes, determine the choice of treatment tactics, as well as the search for new therapeutic approaches.
|
2. Main Body
2.1. Material and Methods of Study
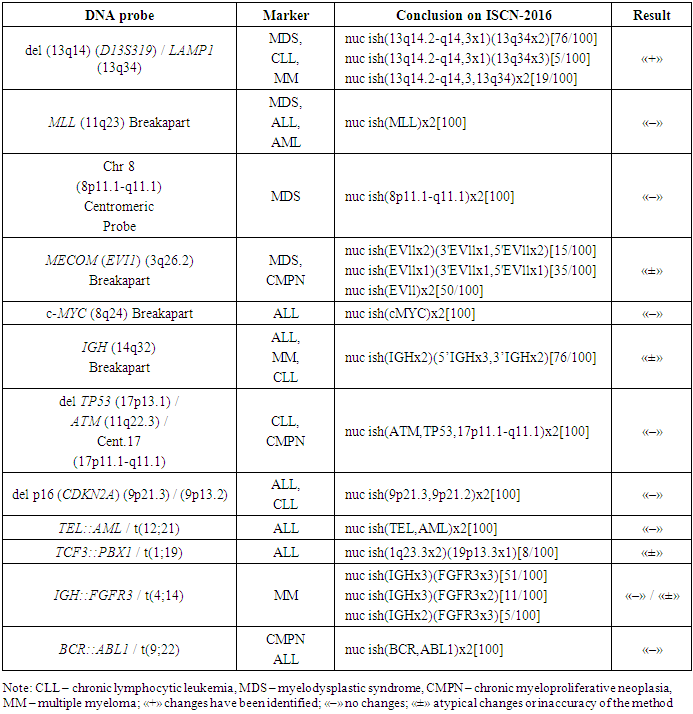
- The patient R.O., born in 1996, was examined and treated at the Republican Specialized Scientific and Practical Medical Center of Hematology of the Ministry of Health of the Republic of Uzbekistan (RSSPMCH of MoH RUz) from October 2021 to October 2022. Diagnostic studies were carried out using generally accepted hematological, biochemical and cytochemical tests.Immunophenotyping of bone marrow cells by flow cytofluorometry (FLOW) was performed on a flow cytofluorimeter «BDFacsLiryc» (USA) using tests to determine CD-markers of progenitor cells, CD-markers of multiple myeloma, and also myeloid, B- and T-linear lymphoid CD-markers. The obtained data about lymphocyte subpopulations were processed in the BDFacsLiryc/FacsSuit application.A conventional cytogenetic study of dividing bone marrow cells was performed using G-banding technology. The analysis of 25 metaphase plates was carried out. For microscopy of cytogenetic slides and the search of metaphase plates an “AxioScope.A1” microscope (Zeiss, Germany) with an “AxioProgRes” video camera was used (magnification during scanning – x 200, during photofixation and analysis – x 1000). The counting of the number, as well as the analysis of morphology and banding of metaphase chromosomes was carried out in the computer application “VideoTestKaryo 3.1”. Chromosomal abnormalities were assessed in accordance with the principles of the International System of Chromosome Nomenclature (ISCN).Molecular cytogenetic analysis of bone marrow cells using fluorescent in situ hybridization technology (interphase FISH) was performed using DNA probes («Diagen», Turkey): «del(13q14)(D13S319) / LAMP1(13q34)», «MLL(11q23) Breakapart», «Chr8(8p11.1-q11.1) Centromeric Probe», «MECOM(EVI1)(3q26.2) Breakapart», «c-MYC(8q24) Breakapart», «IGH(14q32) Breakapart», «del TP53(17p13.1)/ATM(11q22.3) / Cent.17(17p11.1-q11.1)», «delp16(CDKN2A)(9p21.3) / (9p13.2)», «TEL::AML / t(12;21)», «TCF3::PBX1 / t(1;19)», «IGH::FGFR3 / t(4;14)», «BCR::ABL1 / t(9;22)». Hybridization was carried out for 18 hours in automatic mode using the TDH-500 «ALLSHENG» hybridizer (China). The results of hybridization (the number and relative position of fluorescent signals) were analyzed in 100 interphase cell nuclei.
2.2. Results of the Study
- Case DescriptionClinical observation. From anamnesis vitae: R.O. grew up and developed according to his age. Poor state of health and bone pain since October 2021 have been the reason for his request for specialized medical care at the RSSPMCH of the MoH RUz. According to the results of multispiral computed tomography performed in October 2021, rounded foci of osteolytic destruction were detected in the patient, while the level of alkaline phosphatase of blood was 184 units/l. Based on the results of hemogram, myelogram, immunohistochemical analysis and immunophenotyping, acute leukemia and multiple myeloma were excluded. The patient was recommended to consult an oncologist. In April 2022, the patient was examined in a private clinic, where he was given a preliminary diagnosis with the wording "malignant neoplasm of bones of independent multiple localizations" and prescribed treatment (Prednisone, Revolade, transfusion of blood components). The patient received Prednisone therapy (30 mg per day orally) for three months. At first, after the start of treatment, the patient subjectively felt an improvement, but in July 2022 his health status worsened again.In September 2022, the patient again sought help from the RSSPMCH of the MoH RUz. According to the hemogram performed on September 7: hemoglobin – 70,0 g/l, red blood cells – 2,35 x 1012/l, platelets – 4,7 x 109/l, white blood cells – 3,15 x 109/L, rod-shaped neutrophils – 12,0%, segmented neutrophils – 30,0%, lymphocytes – 43,0%, erythrocyte sedimentation rate – 70 mm/h; myelograms: blast cells – 8,0%, lymphocytes – 26,4%, megakaryocytes were not detected. At the same time, immunophenotyping showed the presence of 8,2% of blast cells of B-lymphoid origin in the bone marrow (CD10 – 44%, CD19 – 74%, CD79a – 53%); plasmocytes were not detected.Despite the severity of the condition, the patient refused from hospitalization. However, in October 2022, due to a sharp worsening in well-being, the patient was hospitalized again at the RSSPMCH of the MoH RUz. According to an objective examination, the patient's general condition was assessed as severe with complaints of significant weakness, hemorrhages and nosebleeds, headaches and dizziness, bone pain, up to loss of the ability to move independently. There was no increase in peripheral lymph nodes. The spleen was enlarged (+2.0 cm) and dense. Other indicators of status praesens during the examination were within the limits of the norm.According to the hemogram performed on October 13, 2022: hemoglobin – 61,0 g/L, red blood cells – 2,16 x 1012/L, platelets – 5,0 x 109/L, white blood cells – 9,78 x 109/L, blast cells – 5,0%, normoblasts – 9,0%; myelocytes – 1%, rod-shaped neutrophils – 5,2%, segmented neutrophils – 18,3%, eosinophils – 0,5%, basophils – 0,1% monocytes – 29,9%, lymphocytes – 51,2%, erythrocyte sedimentation rate – 63 mm/h; myelograms: blast cells – 87,6%, a decrease in granulocyte germ, megakaryocytes were not detected. The immunophenotype of the patient's bone marrow cells as of October 13 was represented by the expression of CD34 progenitor cell markers in 89% and antiTdT in 40% of cells, B-linear markers CD10 was detected in 94%, CD19 in 97% and CD79a in 97% of blast cells. On the basis of the data it was concluded that the population of blast cells corresponds to B-ALL.Cytochemical analysis performed in September 2022 and a month later showed a negative reaction to myeloperoxidase; a positive reaction to glycogen in the form of small and large granules was detected in single blast cells.The results of the FISH analysis of bone marrow performed in October 2022 showed (Table 2) that an interstitial deletion of the long arm of chromosome 13 (region 13q14.2-q14.3) was detected in 81% of cell nuclei.
|
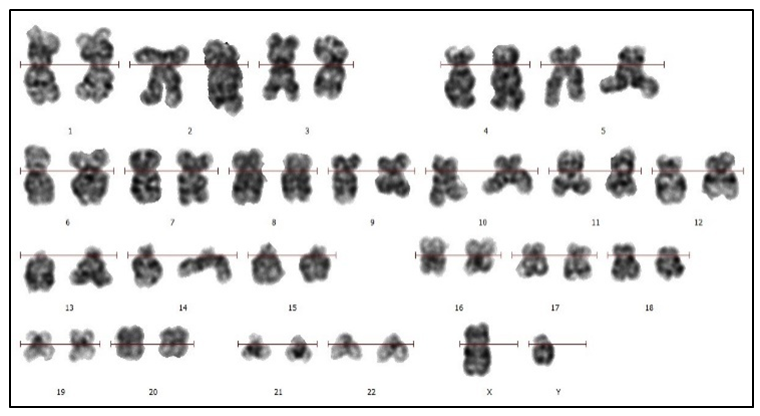 | Figure 1. Karyotyping of leukemic cells of the patient R.O. Chromosomal idiogram (ISCN) performed in the computer application “VideoTestCaryo 3.1”. Karyotype: 46,XY. G-banding. Magnification of х1000 |
 | Figure 2. Karyotyping of leukemic cells of patient R.O. Metaphase plate with hyperdiploid karyotype (51,XY). G- banding. Magnification of х1000 |
3. Discussion
- The described clinical case of a 30-year-old man with Common (BII) ALL with the typical immunophenotype CD10+CD19+CD79a+TdT+CD34+ and hyperdiploid karyotype differed in features that consisted in the development of deep anemia and thrombocytopenia at the onset of the disease, as well as the formation of multiple lytic foci in the bones.Due to the absence of characteristic clinical and laboratory manifestations in the onset of the disease, the diagnosis of clonal neoplastic disease of the hematopoietic system was not made to the patient. The identified foci of bone tissue destruction and impaired musculoskeletal function could indicate metastatic bone damage, and therefore the patient was prescribed an oncologist's consultation. The described bone lesion required biopsy of the osteolytic lesion under the guidance of computed tomography followed by immunohistochemical examination. However, this diagnostic procedure was not performed, since the immunohistochemical method of research is not carried out in the republic, and the export of biomaterial abroad is legally limited.Upon repeated examination at the RSSPMCH of the MoH RUz 11 months after the initial treatment, the hemogram showed signs of inhibition of erythroid and megakaryocytic hematopoiesis germs. The level of blast cells in the patient's blood and bone marrow was not the basis for the diagnosis of acute leukemia, however, the immunophenotype of 8,2% of bone marrow blast cells (CD10+CD19+CD79a+) indicated their belonging to the B-lymphoid line of differentiation. A study of the cellular composition of blood and bone marrow, conducted a month later against the background of deterioration, revealed an increase in the number of lymphocytes and the appearance of blast cells in blood, as well as obvious blastosis (87%) of the bone marrow. The cytochemical analysis data indicated in favor of the lymphoid linearity of the detected systemic blood disease. The results of immunophenotyping confirmed that the patient's blast cell population corresponded to B-ALL.Of the DNA probes used for FISH to identify ALL markers, only a test for a change in the IGH gene was conditionally positive, since the observed pattern of fluorescent signals could indicate both a rearrangement of the gene and the appearance of additional chromosomal material containing IGH, including an additional copy of chromosome 14. At the same time, a FISH analysis test using an additional DNA probe for deletion of the long arm of chromosome 13 (region 13q14.2-q14.3) showed a positive result in more than 80% of interphase cell nuclei.Abnormalities of chromosomal region 13q14 occur in hematological malignancies of all lines and all stages of differentiation and are often found in lymphoid hemoblastoses. Thus, in CLL, del(13q) is detected in 50-60% of cases [5], in multiple myeloma – in 15-24% of cases [6,7,8], with ALL – in 4% of cases [9]. However, the loss of chromosome 13 material was also detected in myeloid neoplasia [10,11].The breakpoints in the loss of chromosome 13 material can vary from 13q12 to 13qter, while ALL is characterized by the involvement of the 13q14 region, in which the retinoblastoma gene (RB1) is located, which is a suppressor gene and is involved in cell cycle control and cell differentiation [10]. There is evidence that with del(13)(q14), not only RB1 is lost, but also neighboring genes, including the DLEU suppressor gene (13q14.2-q14.3) [12]. Deletions affecting the 13q12.2 region in B-ALL have recently been described. These deletions lead to changes in the structure and expression of the CDX2, FLT3 and PAN3 genes located in region 13q12.2. CDX2 is not expressed in hematopoietic cells in the postnatal period of life, but CDX2 expression in cases of B-ALL is 1000 times higher than in other forms of leukemia. The proven involvement of changes in the CDX2, FLT3 and PAN3 genes in oncogenic activation became the basis for the inclusion of patients with CDX2 deregulation by the PAN3 enhancer (13q12.2) into a new separate subgroup of B-ALL [13,3].Deletion of chromosome 13 was detected in patient R.O. by the accessible FISH method using a locus-specific DNA probe, in which the fluorescent signal hybridizes with DNA at the 13q14.2-13q14.3 locus. Analysis of the hybridization result of the probe at the specified locus makes it possible to detect the loss of the fluorochrome-labeled region of the chromosome but does not allow to determine the molecular boundaries of the deleted fragment, which may include a more or less extended region. In this regard, to determine the involvement in the genetic rearrangement of the 13q12.2 locus was not possible.Karyotyping revealed signs of clonal evolution with the presence of two cytogenetic types of leukemic cells: one with a normal karyotype and the second (numerically predominant) with a hyperdiploid karyotype (51,XY). High hyperdiploidy, determined by the presence of 51 or more chromosomes per cell, is often found in tumor cells of ALL patients, and more often in B-ALL [14], as a result of which B-ALL with high hyperdiploidy has been classified by the World Health Organization as a separate subtype [15]. Despite the fact that high hyperdiploidy is associated with a favorable outcome, it has been shown that factors such as the patient's age, white blood cell count, specific trisomies and other factors change its prognostic significance [14]. Hyperdiploidy in B‐ALL is more common in children (25-30% of cases). Much less often, this karyotype anomaly is observed in adults (4-8% of cases), usually accompanied by structural abnormalities [14,15]. It is believed that two or more structural abnormalities combined with hyperdiploidy correlate with the unfavorable survival of adult patients, similar to the survival of patients with a complex karyotype [15].The genetic features of the described B-ALL case were a combination of a hyperdiploid karyotype with a structural rearrangement – deletion of a fragment of the long arm of chromosome 13, including the 13q14 locus. The presence of leukemic clones with normal and hyperdiploid karyotypes in the patient simultaneously suggests secondary origin of hyperdiploidy due to impaired chromosome segregation during cell division. Probably del(13)(q14) is also a secondary genetic event in the pathogenesis of the disease, which arose due to the increase of genomic instability of leukemic cells. Apparently, high hyperdiploidy in combination with del(13)(q14) are not only signs of clonal evolution, but also, taking into account the course and outcome of ALL, factors of unfavorable prognosis.
4. Conclusions
- The presented clinical case of B-ALL was featured by the peculiarities of the course at the onset of the disease, the absence of marker genetic abnormalities and the presence of del(13)(q14), which did not allow to determine precise diagnosis timely. The described case shows that the spectrum of genetic changes in B-ALL with the typical immunophenotype CD10+CD19+CD79a+TdT+CD34+ may include not only recurrent genetic abnormalities that determine the subtypes of the disease, but also other chromosomal rearrangements, the diagnostic and prognostic significance of which will be established in the future as data accumulate.
Conflict of Interest
- The authors declare no conflict of interest.
Funding
- The study was performed without external funding.
Compliance with Patient Rights
- The study was carried out in accordance with the WMA Helsinki Declaration as amended in 2013. Informed consent for publication of data was obtained from relatives of the patient.
Authors’ Contributions
- The authors declare the compliance of their authorship according to the international ICMJE criteria.Yu.Yu. Assesorova – karyotyping examination, evaluation of karyotyping and FISH results, data analysis and interpretation, elaboration of the work conception, reviewing of publications of the article’s theme, article writing;T.R. Alimov– idea of publication, molecular cytogenetic analysis (FISH) examination;I.T. Kodirova– patient management and treatment, clinical and laboratory research material collection, clinical data analysis and interpretation. K.T. Boboev – technical editing, final approval of the version to be published.