-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2025; 15(3): 568-574
doi:10.5923/j.ajmms.20251503.17
Received: Jan. 27, 2025; Accepted: Feb. 20, 2025; Published: Mar. 8, 2025

Metagenomic Analysis of the Nasopharyngeal Microbiota in COVID-19 Patients: Opportunistic and Pathogenic Infections
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLJuamamurodov Sobirjon Tursunboy Oglu1, 2, 3, Nuruzova Zukhra Abdukadirovna4
1Researcher, Laboratory of Molecular Genetics and Cytogenetics Research, Center for Biomedical Technologies, Tashkent Medical Academy, Tashkent, Uzbekistan
2Doctor of the Virology Laboratory of the Chilanzar District, Department of the Tashkent City, Administration of the Tashkent City Administration of the Committee for Sanitary and Epidemiological Welfare and Public Health of Uzbekistan, Tashkent, Uzbekistan
3PhD Student, Department of Microbiology, Virology and Immunology, Tashkent Medical Academy, Tashkent, Uzbekistan
4Ph.D., Professor, Department of Microbiology, Virology and Immunology, Tashkent Medical Academy, Tashkent, Uzbekistan
Copyright © 2025 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Using a metagenomic analysis approach, this study aimed to investigate opportunistic and pathogenic bacteria within the nasopharyngeal microbiota in COVID-19 patients. Create a phylogenetic history using 16S rRNA to determine the genotype of bacterial microflora from nasopharyngeal swab samples, enabling the identification of microbial communities and pathogenic species in COVID-19 patients. Nasopharyngeal swab sample collection and preparation, DNA extraction, amplification of bacterial genetic material (for example, by gene 16S rRNA), metagenomic sequencing (ABI 3500 Genetic analyzer Thermo Fisher Scientific), bioinformatic analysis, microbiota composition assessment, statistical analysis, and comparison of results were used to study the nasopharyngeal microbiota in patients with COVID-19. Fragments were detected in 75% of electrophoresis (n = 27). Detection of fragments indicates the presence of bacteria. When sequencing, pathogenic and opportunistic infections examined Sanger were detected for humans (n=24). These bacteria were detected during sequencing of Bordetella bronchiseptica uradi, Haemophilus influenzae, Chryseobacterium sp, Agrobacterium tumefaciens, Pseudomonas sp, Stenotrophomonas maltophilia, Haemophilus influenzae, Bordetella bronchiseptica uradi, Streptococcus pneumoniae, Staphylococcus epidermidis, Staphylococcus aureus, and Klebsiella pneumoniae. Sequencing results showed that these bacteria were present in the patients' upper respiratory tract. This increases the likelihood of a change in the microflora or the development of additional infections. These bacteria can be associated with various diseases and infections of the respiratory tract. Molecular-genetic and sequencing studies reveal the mechanisms by which COVID-19 can detect secondary bacterial infections in patients and enable the detection of highly human-to-human bacteria that are not detectable in a bacteriological laboratory. These indicators are important in determining treatment strategies for patients and identifying possible bacterial infections.
Keywords: COVID-19, Nasopharyngeal microbiota, Metagenomic assay, Opportunistic pathogens, Bacterial infections
Cite this paper: Juamamurodov Sobirjon Tursunboy Oglu, Nuruzova Zukhra Abdukadirovna, Metagenomic Analysis of the Nasopharyngeal Microbiota in COVID-19 Patients: Opportunistic and Pathogenic Infections, American Journal of Medicine and Medical Sciences, Vol. 15 No. 3, 2025, pp. 568-574. doi: 10.5923/j.ajmms.20251503.17.
Article Outline
1. Introduction
- This study was carried out using a metagenomic analysis approach to investigate opportunistic and pathogenic infections within the nasopharyngeal microbiota in COVID-19 patients. In the study, the genotype of bacterial microflora was determined from nasopharyngeal swab samples, and the presence of opportunistic and pathogenic species was analyzed. With this method, microbiome changes in COVID-19 patients and associations with potential pathogens are identified, which can help explore new possibilities for disease development and treatment.In addition, the study looked at bacterial species present in the nasopharyngeal microbiota and their impact on COVID-19 disease. The species, such as Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria, may vary depending on the health status of the patient. The study also explored how alterations in the nasopharyngeal microbiota play a role in the infection process, especially in COVID-19 patients and what differences are present in mild or severe course of disease.Moreover, the identification and analysis of genetic material in the composition of the microbiota using the approach of metagenomic analysis creates new opportunities in the diagnosis and prognosis of the disease. The technique can also help improve patients' health status and effectively treat COVID-19 infection by manipulating the microbiome using plants or other environmental factors. One of the future goals of the study is to develop new therapies that interact with the microbiota and improve patients' long-term health status.
2. Methods
- This research work was carried out at the Virology laboratory of Chilanzar district of Tashkent city and the Center for Biomedical technologies of the TMA. The object of the study is 36 nasopharyngeal swabs taken from the upper respiratory tract of COVID-19 positive patients admitted to the Republican Special Hospital No. 2 Zangiota in 2021. Of these, the number of males is 14 and females are 22. The ages of the patients ranged from 12 to 74 years.The study used the following methods to study the nasopharyngeal microbiota in COVID-19 patients. Nasopharyngeal swab samples were taken from the patients and kept at −80°C. DNA/RNA was isolated. The obtained samples were processed using special collections and the quality analysis was confirmed by spectrophotometer and gel electrophoresis. In the latter stage, the V3–V4 regions of the 16S rRNA gene were sequenced and the composition of the bacterial microbiota was determined. The sequences data were collected in ab1 format and bacterial species were identified using the U.S. Blast database.
3. Result
- 36 nasopharyngeal smears were taken from patients of the Republican Special Infectious Diseases Hospital No. 2 Zangiota. Samples were collected using a special swab on the surface of the mucous membrane of the upper respiratory tract. Adults and children were involved in this process. During the sampling process, the patient was taken with caution if he or she had recently undergone a nasal injury or surgery. The procedure was completed by applying personal protective equipment (gown, non-sterile gloves, N95 mask and face shield). Prior to the start of work, all test tubes were marked with the appropriate labels and request forms were filled out. The risk of contamination was reduced by strictly adhering to the SHV wearing procedures. "RNase-free" and "DNase-free" sterile solutions were utilized for specimen. In this study, in the process of NK isolation, a package "MagSorbNA", manufactured by ROSSA, was used. This kit is designed for the efficient separation of RNA and DNA. From the samples taken for testing, n=36 samples were sampled by random selection. DNA/RNA was isolated from the samples taken and each was measured with a sphectophotometer. The concentration and purity of the extracted DNA/RNA were evaluated using a spectrophotometer at 260/280 and 260/230. The mean value (64.56) and standard deviation (±16.93), the A260/A280 ratio of 1.8–2.0 were determined to determine the main variables. Two targets in the viral genome (ORF1ab and N gene) and human genome DNA were detected using the RT-PCR method to analyze SARS-CoV-2 RNA. The ROSSAmed COVID-19 RT-PCR kit was used in this study. In addition, the package for internal control of PCR was used to control the quality of biological material extraction and the correct performance of the analysis process. The following are the primers and probes corresponding to the ORF1ab and N genes used to detect the COVID-19 virus in our kit.1. ORF1ab birth:F-primer: 5’-CCCTGTGGGTTTTACACTTAA-3’R-primer: 5’-ACGATTGTGCATCAGCTGA-3’Zond: 5’-FAM-CCGTCTGCGGTATGTGGAAAGGTTATGG-BHQ1-3’2. N gene:F-primer: 5’-GGGGAACTTCTCCTGCTAGAAT-3’R-primer: 5’-CAGACATTTTGCTCTCAAGCTG-3’Zond: 5’-FAM-TTGCTGCTGCTTGACAGATT-TAMRA-3’In real-time RT-PCR analyses, amplification of ORF1ab and N genes was observed in the HEX channel, while human genome DNA was observed in the FAM channel. These HEX and FAM channels are optical detectors used for the detection of fluorescent signals in real-time PCR amplification, allowing for monitoring the dynamics of the amplification process. This approach provides high reliability and repeatability in the detection process of SARS-CoV-2.To use the device, the amplifier was programmed as follows in accordance with the instructions. The RT-PCR protocol for the detection of SARS-CoV-2 RNA was performed as follows. Reverse transcription took 10 min at 55°C. Initial denaturation was performed for 15 min at 95°C. The denaturation phases of 95°C, (15 sec), otjig 60°C, (30 sec), elongation 67°C, (15 sec) were repeated for 5 primary cycles and 35 primary cycles. Detection was performed at 60°C via HEX and FAM channels. We programmed and amplified the device according to these instructions, according to the location of the solutions in amplification. At the end of the experiment, the results obtained were saved by the device in a file format. Before proceeding the analysis, the device settings were adjusted according to the manufacturer's instructions. The following parameters were obtained for the analysis: Selection of method – "Forgego Method (St)" was selected for the device and the values of the required threshold parameters were entered. Logarithmic Scale – For the visual analysis of the results, logarithmic measurement was used, which helped to identify the linear growth portion of the amplification curve. Boundary Line Settings – To determine the boundary line crossing on the grow portion of the amplification curve, the boundary line has been manually adjusted to the required size. When analyzing the results, it was observed whether or not there is an intersection of fluorescence curve analysis – Amplification curve with boundary line. This was confirmed by the presence or absence of the "Ct" (Threshold value) indicated in the corresponding column of results. Ct Values – The values were analyzed and verified for consistency with the intended values for the control samples. The presence of a threshold value confirmed the presence of an amplified gene or target RNA. Interpretation of results – if Ct values are available, it indicates that the amplification process was successful and the presence of targeted nucleic acids in the sample. In cases where a Ct value is not observed, it means that the sample does not contain the RNA or DNA required for amplification or that their concentration is likely to be below the detection limit.In terms of device and analysis methodology, all parameters were implemented under control. Results were analyzed based on boundary values in the logarithmic part of the amplification curve. The Ct values presented were consistent with the control samples, confirming that the analysis was performed correctly. This guarantees the reliability of the analysis process and the fact that the results obtained accurately reflect the biological data. The ORFab and N gene were detected between 20 and 31 cycles. These were shown as amplified targets during the PCR process. According to the kit manual, patients up to <30 cycles were taken in the HEX channel. The <30 cycle result shows that the amplification process was performed with high precision. This cycle count ensures the accuracy of the amplification process and high safety reliability. Working with fewer cycles ensures that our work is accurate and reliable (Figure 1).
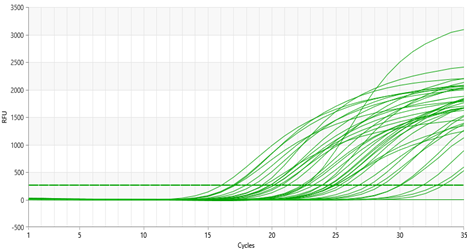 | Figure 1. ORF1ab and N gene in the PCR HEX channel |
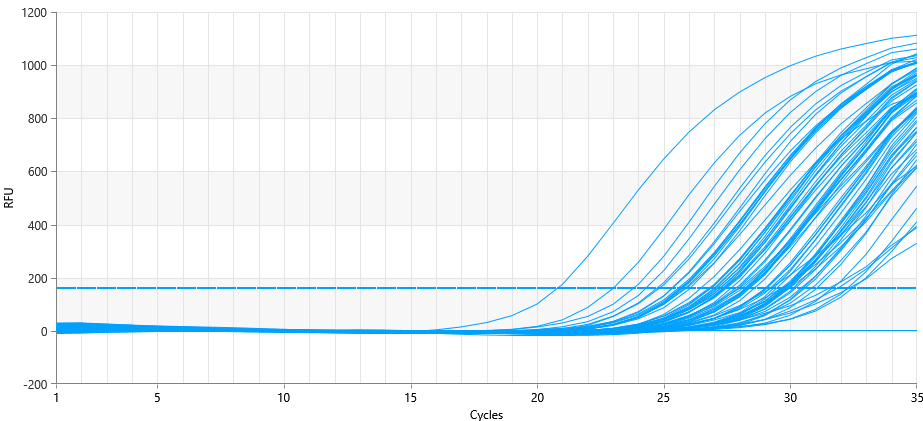 | Figure 2. Internal control in the PCR FAM channel |
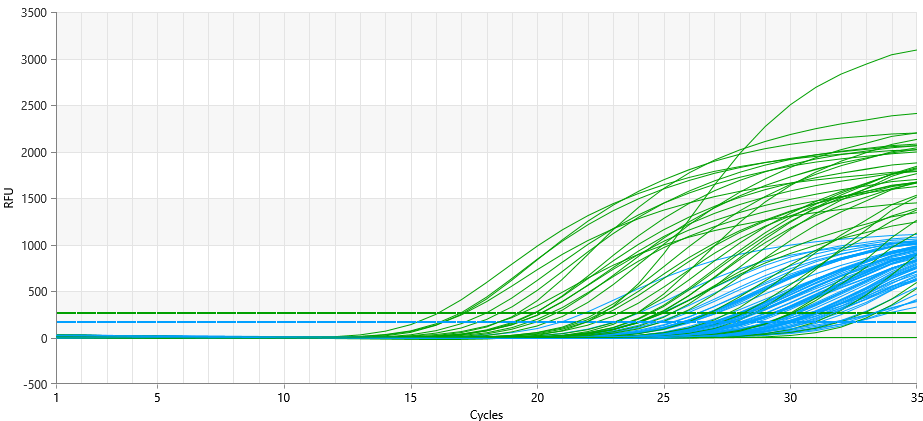 | Figure 3. FAM and HEX channel at PCR |
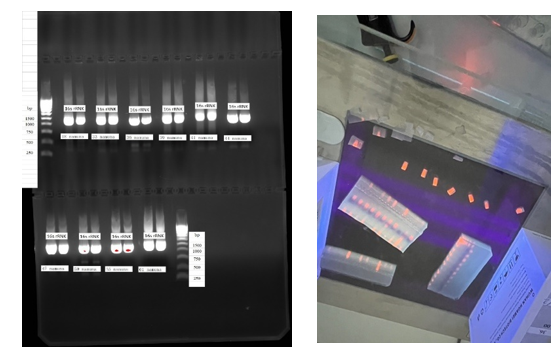 | Figure 4. 16S ribosomal RNA gene: Visualization and cross-cutting process in agarus electrophoresis |
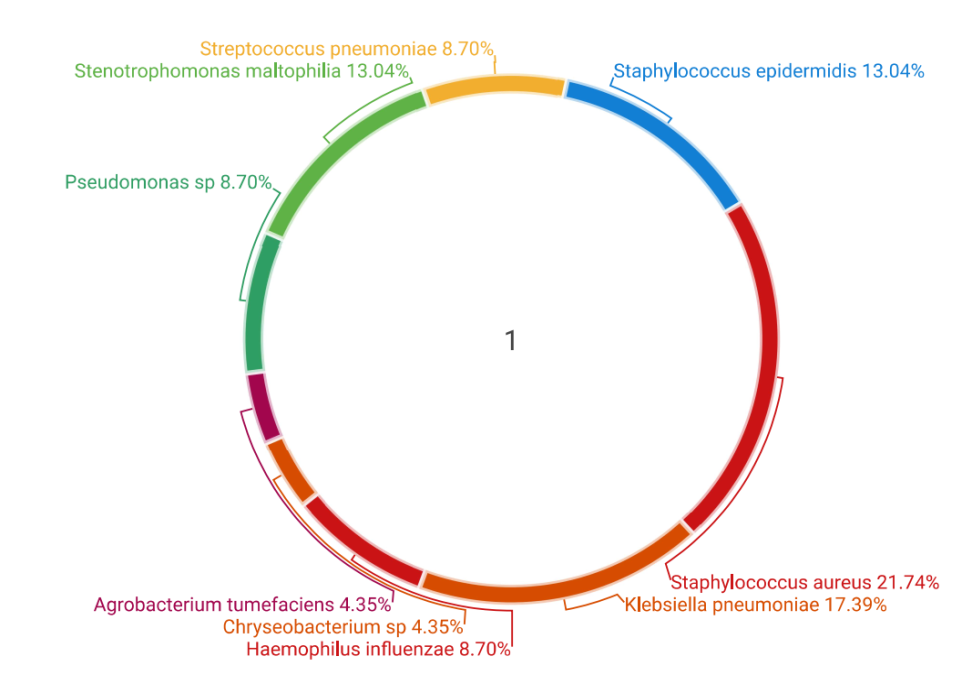 | Figure 5. Bacterial microflora of the upper respiratory tract in COVID-19 patients |
 | Figure 6. The result of sequencing of polyvalent bacteria |
4. Discussion
- Our knowledge of bacterial coinfections and their clinical significance has changed significantly during the COVID-19 pandemic. A Peruvian hospital showed that the prevalence of bacterial pathogens in patients with COVID-19 was 41%. This indicator corresponds to the data in the range of 0.6-50% given in the literature. [1]One of the most important findings of our study is that the traditional pneumonia causative agent (Staphylococcus aureus). This result indicates the need to rethink empirical antibacterial and antiviral therapy strategies. Even in a study in China, co-infection of SARS-CoV-2 and the influenza virus was recorded in only 0.4% of cases. [2]Two factors may have contributed to the high prevalence of bacterial pathogens. First, most patients had already taken azithromycin prior to hospitalization, which may have changed normal microflora and increased susceptibility to colonization. Second, the molecular diagnostic methods used are highly sensitive to traditional cultures. [3] [4]The important point is that it is possible to determine whether the microorganisms identified by molecular methods are a real pathogen or a simple colonizer only on the basis of clinical criteria. Metagenomic sequencing studies have shown that certain bacteria, such as S. agalactiae, may increase ACE-2 receptor expression and increase susceptibility to SARS-CoV-2 infection. [5]Key limitations of our study include:1. Widely used antibiotics before hospitalization2. Relatively small sample size3. Not a complete analysis of antibiotic use in the hospitalFor future research, it is recommended to use metagenomic sequencing techniques, study microbiome changes in depth, and conduct prospective follow-up in an outpatient setting. This will provide a better understanding of the role and importance of bacterial coinfections in patients with COVID-19.
5. Conclusions
- These indicators are important in determining treatment strategies for patients and identifying possible bacterial infections. Results should be carefully analyzed to monitor infections and take timely action.The study helped identify changes in the nasopharyngeal microbiota in COVID-19 patients and assess the risk of secondary bacterial infections. The results showed that the metagenomic analysis approach expands the diagnostic capabilities.
ACKNOWLEDGEMENTS
- We express our sincere gratitude to the staff of the Center for Biomedical technologies of the Tashkent Medical Academy and the specialists of the Committee for Sanitary and Epidemiological Welfare who helped to implement this research.
Data Availability Statement
- All data collected during the research will be retained in accordance with the privacy policy and may be made available at the request of the researchers.
Author's Contributions
- The main contribution to the design and conduct of the research were S. Juamamurodov and Z. Nuruzova. Bioinformatic analyses and recording works were performed by S. Juamamurodov.
Conflict of Interest
- The authors declare that there are no conflicts of interest with respect to this study.