-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2024; 14(11): 2944-2947
doi:10.5923/j.ajmms.20241411.58

Microcephaly in Children: Early Signs and Diagnosis
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAbdullaeva Muazzamkhon Ilkhomidinovna, Kuchkarova Odinakhon Bakhromjonovna
Department of Neurology, Andijan State Medical Institute, Andijan, Uzbekistan
Copyright © 2024 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Microcephaly is a serious neurological condition manifested as a reduced size of the child's head compared to normal values for a given age and sex. It may be caused by underdevelopment of the brain during pregnancy or its slow growth after birth. The condition can affect the child's overall development and lead to a variety of neurological problems, so early detection and diagnosis are crucial for timely intervention and support. This article discusses key aspects of the early signs of microcephaly and diagnostic methods, which can assist parents and health professionals in the rapid identification and appropriate response to the condition.
Keywords: Microcephaly, Developmental delay, Neurological disorders, Causes of microcephaly, Genetic abnormalities, Zika virus, Perinatal complications, Seizures
Cite this paper: Abdullaeva Muazzamkhon Ilkhomidinovna, Kuchkarova Odinakhon Bakhromjonovna, Microcephaly in Children: Early Signs and Diagnosis, American Journal of Medicine and Medical Sciences, Vol. 14 No. 11, 2024, pp. 2944-2947. doi: 10.5923/j.ajmms.20241411.58.
1. Introduction
- Microcephaly is a neurological condition in which a person has a head smaller in size than peers of the same sex and age, often due to abnormal brain development. In pediatrics, 1 baby in 1,000 is noted to have microcephaly of the brain. This disorder is characterized by a decrease in the skull and brain mass of the child. The body, however, develops absolutely normal, without any abnormalities [1]. The consequence of microcephaly is various mental abnormalities. It can be of various forms, ranging from imbecility to all sorts of forms of idiocy. It is impossible to be completely cured of this disease. A person diagnosed with microcephaly can live up to 15 years, under favorable conditions, life expectancy reaches 30 years. Microcephaly in a certain percentage of sick children caused mutations at the gene level. In total, there are two types of disease: primary and secondary. More chances to become more independent and adapt to the performance of any actions have a person belonging to the first group [2]. Determine the development of microcephaly can be independently, the first sign - the circumference of the head. Microcephaly is a reduction in the size of the skull due to underdevelopment of the brain, clinically accompanied by mental retardation and neurological disorders. Microcephaly is divided into hereditary, embryopathic and syndromic (as a syndrome of almost all chromosomal abberations and in some metabolic diseases). Frequency of occurrence 1.6 cases per 1000 newborns [3].True microcephaly is understood to be an inherited form. Both sexes are affected with equal frequency. Familial cases have been described. Microcephaly is transmitted by autosomal recessive and sex-linked inheritance. Phenotypically healthy relatives show a decrease in the size of the skull, low intelligence. In the pedigree of such patients there are often cases of oligophrenia, seizures, etc. The recessive type of inheritance is also confirmed by a large percentage of consanguineous marriages [4]. Many factors are important in the occurrence of embryopathic microcephaly: infections (influenza, toxoplasmosis, rubella), intoxication (alcoholism, occupational hazards), violation of vitamin balance. The cause of microcephaly may also be hypoxia, violation of mineral metabolism (calcium and phosphorus). The brain in microcephaly is reduced in size. The ratio of brain weight to body weight is 1:33 (with microcephaly 1:100). The cortex of the large hemispheres, which is underdeveloped, is most affected. Other parts of the brain also have an irregular structure. Especially large changes are noted in the frontal lobes: they are much smaller than normal, gyrus are few, flattened, smoothed. Often there are malformations of the corpus callosum. In the occipital lobes, as a rule, there are fewer changes [5]. The proportion of cortex and trunk sections is disturbed. In the norm, the mass of the cerebellum is relative to the brain mass 1:8. In microcephaly, this value is increased. The cerebellum often remains uncovered by the large hemispheres. The brain stem is poorly myelinated as in premature infants. Microcephaly is often accompanied by underdevelopment of the vascular system, especially in the middle cerebral artery basin. Nerve cells are reduced in size, with small outgrowths, with impaired synaptic communication [6]. The clinical picture of microcephaly depends on the stage of fetal development in which a particular factor acted. In some cases, the diagnosis of microcephaly can be made at birth or in the first few months of life on the basis of a tendency to a slow increase in the size of the head and the disproportion between the rate of increase of the head and the circumference of the chest [7].The leading symptom is a disproportion between the cerebral and facial parts of the skull, between the head and trunk. Very characteristic appearance of the patient: the head is narrowed to the top, the forehead is low, sloping, protruding supraorbital arches, eyelashes are very long, thick, ears are large, protruding, asymmetrical, low, teeth are large, sparse, often carious, the palate is high, narrow. The growth of the skull is more in length, and its height does not change much during life.The main place in the clinical picture are symptoms of intellectual defect (idiocy, imbecility, less often debility). In addition to mental disorders, there are changes in the nervous system: impaired function of the oculomotor nerves, changes in muscle tone, often spastic paresis, seizures. Diagnosis of microcephaly is based on clinical symptoms, biochemical tests, radiologic and cytologic studies, the results of ultrasound diagnosis of the brain and genetic counseling. The differential diagnosis of microcephaly is made with microcrania.Craniostenosis leads to a reduction in the size of the head, while there is no disproportion of the facial and cerebral parts of the skull. Craniostenosis is characterized by increased intracranial pressure, headaches, strabismus, pyramidal signs, and craniogram changes.Primary (hereditary) microcephaly: Mendelian inheritance or association with a genetic syndrome: o MUPI: head circumference > three standard deviations below the mean, simplified pattern of gyrus architectonics, shallow furrow depth o Microlissencephaly: head circumference > three standard deviations below the mean, pachy or agyria. Secondary (nonhereditary) MCF: effect of a toxic factor on brain growth during fetal, neonatal, or infancy period.General characteristics of microcephaly:- Best diagnostic criteria: o Craniofacial disproportion, overlapping sutures, simplified gyrus architectonics, shallow depth of furrows o Visualization signs depend on the cause of microcephaly o In more severe forms of microcephaly, simplification of the pattern of gyrus architectonics is noted o Microcephaly is classified by gene mutation (if known); and: - Relative size of supratentorial/infratentorial structures - Supratentorial anomalies (polymicrogyria, heterotopia, callosal anomalies) - Combination with developmental anomalies outside the CNS: Musculoskeletal malformations, heart, gastrointestinal malformations, etc. - Visualization: depends on the cause of microcephaly: o Primary (hereditary) microcephaly: prenatal or postnatal, often associated with developmental delay (DD) - Prenatal microcephaly: associated with severe microcephaly (< 3 standard deviations from the mean) at birth - Postnatal microcephaly: Small normal head size at birth, drop in percentiles (10% to < 1%) in infancy o Secondary (nonhereditary) microcephaly: - Hypoxic-ischemic encephalopathy (HIE) ± loss of cortical, white matter or basal ganglia volume - TORCH infections: Ca++, white matter abnormalities, neuronal migration abnormalities, germinolytic cysts - Pediatric concussion syndrome: encephalomalacia, chronic subdural hematomas ± ruptured brain parenchyma.- Lateral radiographs, review CT scans, or sagittal MR tomograms: craniofacial disproportion: o Normal craniofacial ratios: ↓ premature (5:1), preterm (4:1), two years (3:1), three years (2.5:1), 12 years (2:1), adults (1.5:1).Radiologic signs of microcephaly: - Radiography: o ↓ craniofacial relationship, slanted forehead, convergence or overlap of the sutures of the cranial vault bones.3. CT for microcephaly: - Contrast-free CT: o Small skull vault: close convergence of sutures, overlap ± secondary craniosynostosis o Ca++ in TORCH and pseudo-TORCH syndromes o Cortical surface: normal ↔ simplified architectonics ↔ migration anomalies ↔ microlissencephaly ↔ microlissencephaly (Figure 1,2).
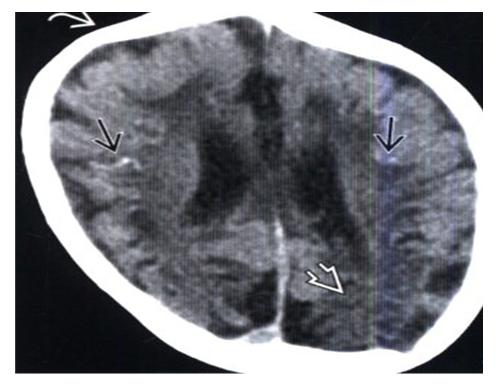 | Figure 1. Contrast-free CT scan, axial slice: a neonate with microcephaly who underwent rapid labor due to placental abruption shows scattered foci of subcortical calcification combined with cortical atrophy and overlap of the right coronal suture, indicating microencephaly |
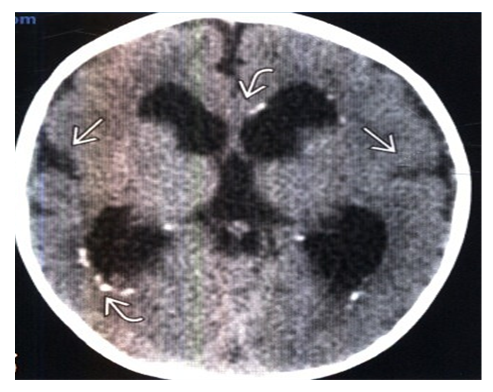 | Figure 2. Contrast-free CT scan, axial slice: a newborn with microcephaly and congenital CMV infection shows scattered foci of periventricu-lar calcification. Note the smoothing of the sylvian furrows combined with simplification of the pattern of gyrus architectonics (polymicrogyria was detected on MRI) |
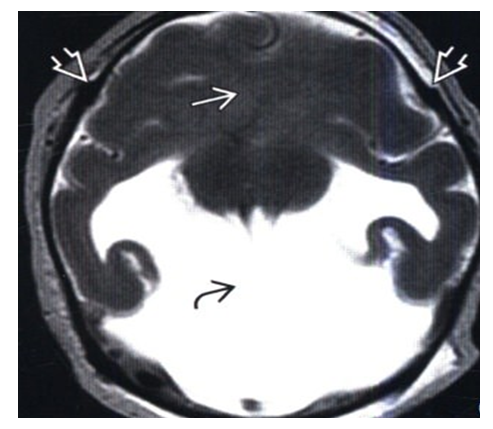 | Figure 3. MRI, T2-VI, axial slice: extended hemispheric cystic encephalomalacia is detected in a newborn with microcephaly due to intrauterine infarction in the middle cerebral artery blood supply basin |
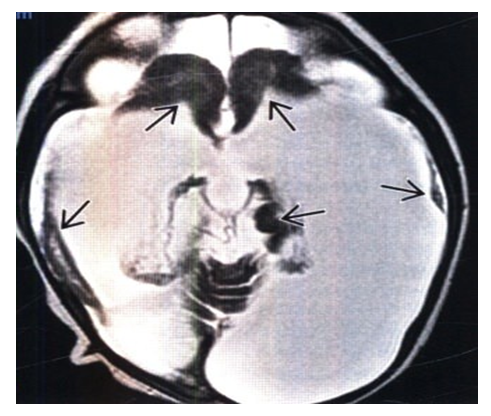 | Figure 4. MPT, T2-VI, axial slice: in a neonate with severe microcephaly and hydranencephaly, it is noted that the area of absent brain tissue corresponds to the blood supply basins of the internal carotid arteries. Note the small islands of residual brain tissue belonging to the frontal and temporal lobes |
2. Conclusions
- Thus, it is possible to use ultrasound diagnosis of cranial bone sutures to assess whether there is no suture fusion and to make a differential diagnosis between the two conditions. It is performed in the first few months of life. Drug therapy: stimulating, sedative, anticonvulsant, dehydration. Of importance are: massage, physical therapy, educational activities, labor adaptation. Children with a head size less than normal or on the border of the norm and with a tendency to disproportion of the head and trunk, brain and facial parts are subject to dispensary observation of neurologist, pediatrician, with mandatory monthly measurement of the circumference of the head, chest, height, body weight, during the first year of life. In the second and third year of a child's life, the examination is carried out once every 6 months.Currently, specialized nurseries, kindergartens, boarding schools are organized for children with lesions of the nervous system. Corrective measures significantly improve the social adaptation of such children.