-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2024; 14(11): 2707-2710
doi:10.5923/j.ajmms.20241411.05
Received: Oct. 15, 2024; Accepted: Nov. 5, 2024; Published: Nov. 7, 2024

Results of Molecular-Genetic Research on ABCA4 Gene Mutations in Patients with Hereditary Retinopathy in Uzbekistan
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLOchiliev M. B.1, Oralov B. A.2, Bilalov E. N.3, Narzikulova K. I.3
1Assistant, Department of Ophthalmology, Termez Branch of Tashkent Medical Academy, Uzbekistan
2Doctor of Philosophy Medical Sciences (Phd), Department of Ophthalmology, Tashkent Medical Academy, Uzbekistan
3Doctor of Medical Sciences, Department of Ophthalmology, Tashkent Medical Academy, Uzbekistan
Copyright © 2024 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

The increasing incidence of hereditary retinopathy, partly due to traditions of consanguineous marriages, has made this disease a scientifically and practically significant concern. The genetic basis of retinopathy is tied to the pathogenic significance of genes involved in the metabolism of visual enzymes, such as ABCA4. Objective. To conduct a molecular-genetic analysis of ABCA4 gene mutations in patients with hereditary retinopathy in Uzbekistan. Materials and Methods. The study included peripheral blood samples from 22 patients (13 men and 9 women) clinically diagnosed with hereditary retinopathy. DNA was extracted from nucleated blood cells, and PCR amplification was performed. We designed and synthesized five pairs of oligonucleotide primers for amplifying exons 12-13, 17, 21-22, 28-29, and 41-43. Subsequently, these exons and the associated introns were sequenced using the Sanger method. Results. Based on the study results, a total of 77 mutations were observed across five categories among the patients. The patient with the highest number of alterations, Patient 2, exhibited 6 mutations; Patients 3, 5, 6, and 12 each showed 5 mutations; Patients 7, 11, 13, 14, 15, 16, 17, 18, and 19 had 4 mutations each; while others showed 3 or 1 mutation. Among these, 34 were splice-site mutations, 21 polymorphisms, 18 deletions, 2 synonymous, and 2 missense mutations. Conclusions. The results of the genetic research indicate that hereditary diseases in patients with retinitis pigmentosa (RP) are closely related, with the majority of patients exhibiting an autosomal-recessive inheritance pattern.
Keywords: Retinitis pigmentosa, ABCA4, Gene, Molecular-genetic analysis
Cite this paper: Ochiliev M. B., Oralov B. A., Bilalov E. N., Narzikulova K. I., Results of Molecular-Genetic Research on ABCA4 Gene Mutations in Patients with Hereditary Retinopathy in Uzbekistan, American Journal of Medicine and Medical Sciences, Vol. 14 No. 11, 2024, pp. 2707-2710. doi: 10.5923/j.ajmms.20241411.05.
1. Introduction
- Hereditary retinal diseases affect 1 in 4000 of the global population and are considered hereditary disorders caused by over 3000 different gene mutations. These diseases arise when mutations in specific genes result in nucleotide substitutions or deletions, leading to the synthesis of defective proteins. These mutagenic proteins fail to perform their functions, causing altered clinical symptoms to appear at the phenotypic level. Notably, some of the genes associated with these disorders, such as PRPH2 (chromosome 6), IMPDH1 and RP9 (chromosome 7), and RP1 (chromosome 8), follow an autosomal dominant inheritance pattern; CRB1 (chromosome 1) and SPATA7 (chromosome 14) as well as ABCA4 (chromosome 1) follow an autosomal recessive pattern; and RP2and RPGR are X-linked. This highlights the heterogeneity of the disease. Among these mutagenic genes, ABCA4is one of the most studied genes associated with hereditary retinal diseases. The ABCA4 gene is part of the ATP-binding cassette (ABC) family, which comprises seven subfamilies (ABC1, MDR/TAP, MRP, ALD, OABP, GCN20, White). The ABCA4 gene produces the ABCR protein, which facilitates the transport of trans-retinal aldehyde (atRAL) across the photoreceptor cell membrane. Mutations in this gene are known to cause Stargardt disease, cone-rod dystrophy, cone dystrophy, retinitis pigmentosa, and age-related macular dystrophy. In 2000, a model proposed by Magueri et al. classified ABCA4 mutations into three categories: mild, intermediate, and severe, explaining different phenotypic spectrums based on various combinations of these variants. According to this hypothesis, complex heterozygous combinations of mild and severe variants lead to STGD1; moderate and severe variants cause cone-rod dystrophy, while severe variants on both chromosomes result in retinitis pigmentosa . The phenotypic signs of these genetic mutations often appear early and are associated with lipofuscin accumulation in the retinal pigment epithelium and photoreceptors. The initial symptoms of RP include difficulty adapting to darkness, and nyctalopia, and in some cases, a narrowing of the mid-peripheral visual field, with potential anatomical abnormalities in the central retina. Many patients retain residual macular function, which allows for light perception or a limited peripheral visual field, typically on the temporal side. Photopsia is common but often overlooked in clinical settings. Lu Y. (2021) explains that photopsia arises due to the absence of afferent nerve impulses in response to photoreceptor degeneration or as a result of spontaneous signals due to retinal remodeling. Photopsia appears in the early stages of RP, and in advanced stages, patients may experience vivid visual hallucinations corresponding to Charles Bonnet syndrome, as well as symptoms like photophobia and dyschromatopsia.Several subtypes of RP have been identified, among which the pericentral type is considered atypical, characterized by the preservation of the outer peripheral vision in patients. In this subtype, pigmentation zones resembling bone spicules undergo atrophy in the inner and outer regions of the vascular arcades, resulting in ring scotomas and subnormal full-field ERG results. Such cases were observed in 43 patients by Comander J. (2017) [3], and in 28 patients by Matsui R. (2015) [4]. Information about the dominant heterogeneity of ABCA4 inheritance also exists, showing that the disease pathogenesis may be influenced by hypomorphic alleles [5]. The discovery of a link between complex alleles in ABCA4 and the pathogenesis of hereditary retinal diseases has led to conducting molecular-genetic testing of this gene's mutations in patients in Uzbekistan.Objective. To conduct a molecular-genetic analysis of ABCA4 gene mutations in patients with hereditary retinopathy in Uzbekistan.
2. Materials and Methods
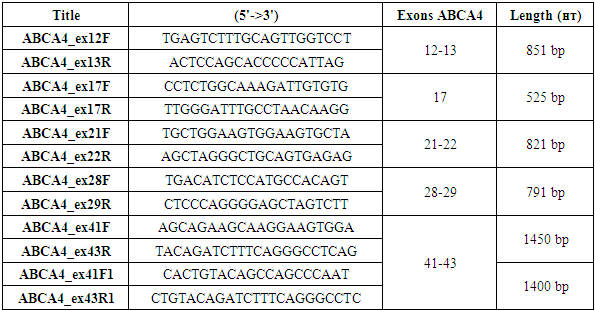
- The study materials consisted of peripheral blood samples from 22 patients (13 men, 9 women) clinically diagnosed with hereditary retinopathy. DNA was extracted from nucleated blood cells, and PCR amplification was performed. For amplification of exons 12-13, 17, 21-22, 28-29, and 41-43, five pairs of oligonucleotide primers were designed and synthesized (Table 1). Subsequently, all these exons and associated introns were sequenced using the Sanger method.
|
3. Results of the Study
- Based on the obtained analysis results, the potential kinship among patients was assessed, and they were divided into three groups. Group 1 included patients 2, 3, 5, 6, 7, 11, 12, 13, 14, 15, 17, 18, and 19, who shared common mutations in introns 12 and 28. All of these patients had the mutation 1761-55G (IVS13-55G>A) in intron 12, and patients 2, 3, 5, 6, 7, and 10-22 showed mutations IVS28 299C>T, IVS28 del334G, and IVS28 337A>G in intron 28. The similarities in mutations observed in these introns indicate close kinship and a common genetic lineage among them. These mutations led to aberrant splicing of mRNA. Group 2 included patients 4, 8, 9, and 16, who only had the mutation 1761-55G (IVS13-55G>A) in intron 12. The presence of a single mutation suggests a more distant kinship among these patients. Group 3 consisted of three patients with specific mutations. Patient 2 had a unique missense mutation c.1699G>A (p.V567M), patient 5 had the missense mutation c.4283C>T in exon 29, and patient 6 had a synonymous mutation 4203C>A (p.P1401P) in exon 28. These specific mutations suggest that these patients belong to distinct genetic lineages.The mutated regions of the ABCA4 gene are shown in Figure 1. Each peak in the chromatogram represents different nucleotides, with four colors indicating adenine (A), cytosine (C), guanine (G), and thymine (T). The red arrow highlights the changed nucleotide peak, indicating the position of the mutation. In this figure, the mutation in the DNA is labeled as c.1699G>A, showing a guanine (G) to adenine (A) change at position 1699, which, when translated to the protein, causes a substitution of valine (V) with methionine (M) at position 567 of the p.V567M protein.
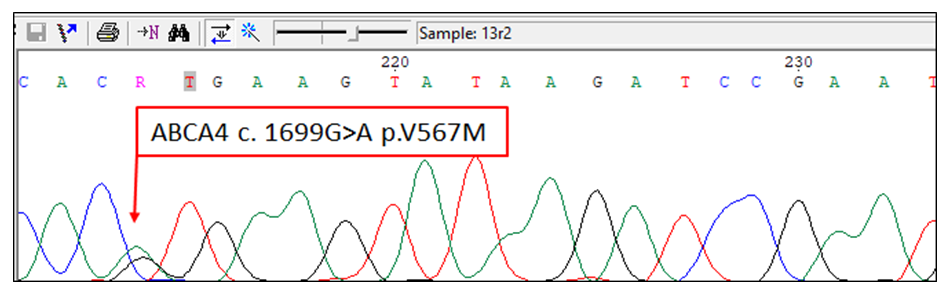 | Figure 1. Sequencing Chromatogram of Mutated Regions in the ABCA4 Gene of Patient 2 |
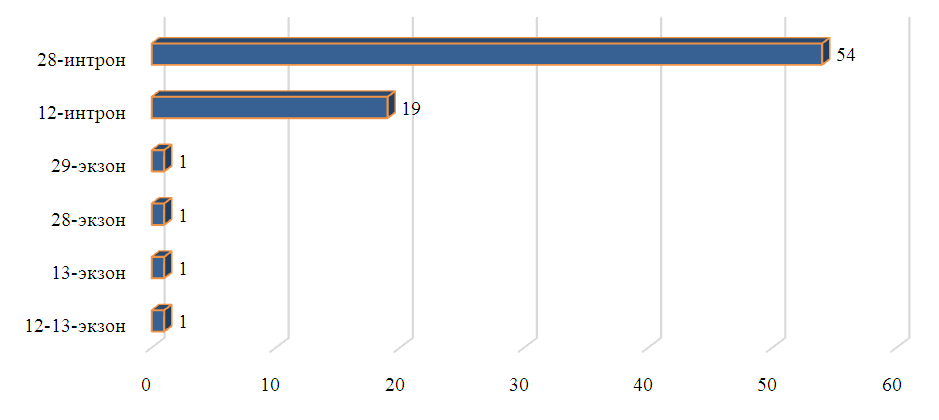 | Figure 2. Frequency of mutations in exons and introns (n = number of occurrences) |
4. Conclusions
- The results of the genetic research indicate that hereditary conditions in patients suffering from retinitis pigmentosa (RP) are closely related, with a significant portion showing autosomal recessive inheritance. The analysis revealed a total of 34 splice mutations (44.2%), 21 polymorphisms (27.3%), 18 deletions (23.4%), and 2 synonymous mutations (2.6%). Among the mutations located in the ABCA4 gene, the most dangerous ones were found in introns 12 and 28.