-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2024; 14(10): 2632-2640
doi:10.5923/j.ajmms.20241410.39
Received: Oct. 5, 2024; Accepted: Oct. 22, 2024; Published: Oct. 24, 2024

Unraveling the Genetic Determinants of Early Hypertension Onset in Young Adults Using Mendelian Randomization
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLDilorom Abidova
Republican Specialized Scientific Practical Medical Center of Cardiology, Tashkent, Uzbekistan
Correspondence to: Dilorom Abidova, Republican Specialized Scientific Practical Medical Center of Cardiology, Tashkent, Uzbekistan.
Copyright © 2024 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

This study aimed to elucidate the genetic factors contributing to early-onset hypertension in young adults using Mendelian Randomization (MR) methods. The hypothesis was that specific genetic polymorphisms play a causal role in the development of hypertension in this population.
Keywords: Early-Onset Hypertension, Mendelian Randomization, Genetic Polymorphisms, Causal Relationships, CYP11B2, ADD1
Cite this paper: Dilorom Abidova, Unraveling the Genetic Determinants of Early Hypertension Onset in Young Adults Using Mendelian Randomization, American Journal of Medicine and Medical Sciences, Vol. 14 No. 10, 2024, pp. 2632-2640. doi: 10.5923/j.ajmms.20241410.39.
Article Outline
1. Introduction
- Hypertension is a major risk factor for cardiovascular diseases and is increasingly recognized as a global health challenge, especially when it presents at an early age. Early-onset hypertension, defined as hypertension diagnosed before the age of 35, affects approximately 5% of the young adult population and is associated with a higher lifetime risk of complications such as stroke, myocardial infarction, and kidney failure. The prevalence of early-onset hypertension has been rising, driven by a combination of genetic, environmental, and lifestyle factors, including obesity, physical inactivity, and high salt intake. Identifying the underlying causes of early-onset hypertension is crucial to prevent its progression and mitigate its long-term health consequences.Genetic factors play a significant role in the development of hypertension, with genome-wide association studies (GWAS) identifying numerous loci associated with blood pressure regulation. Polymorphisms in genes such as ADD1, CYP11B2, and ACE have been implicated in the pathogenesis of hypertension. However, the exact causal pathways through which these genetic variants contribute to early-onset hypertension remain unclear. Understanding the genetic determinants of this condition can provide insights into its etiology and offer opportunities for personalized prevention and treatment strategies.Mendelian Randomization (MR) is a powerful method that leverages genetic variants as instrumental variables to assess causal relationships between exposures and outcomes. By using genetic variants that are randomly assigned at conception, MR minimizes confounding and reverse causality, allowing for more reliable causal inference. This approach has been increasingly applied in cardiovascular research to uncover the genetic factors underlying various conditions, including hypertension.In this study, we aim to investigate the causal relationship between specific genetic polymorphisms and the risk of early-onset hypertension in young adults using Mendelian Randomization. By analyzing data from 450 hypertensive patients aged 18-35, we focus on polymorphisms in the ADD1, CYP11B2, and ACE genes, which have been previously associated with blood pressure regulation. Additionally, we explore the potential gene-environment interactions that may amplify the genetic risk of hypertension in this population, particularly in the context of modifiable lifestyle factors such as salt intake.Through this research, we seek to provide robust evidence of the genetic determinants of early-onset hypertension and identify potential biomarkers for risk stratification. The findings of this study could pave the way for personalized interventions that target individuals with a genetic predisposition to hypertension, ultimately reducing the burden of this condition in young adults. Hypertension (HTN) is a frequent condition with multifactorial cause in which genetic, fetal and environmental factors interact to result in the final phenotype of blood pressure (BP) elevation. Relevance in terms of cardiovascular and renal risk has been demonstrated in multiple epidemiological and intervention studies being one of the most powerful markers for survival. Concerned with the importance in public health and clinical settings, boosted a large body on research in to identify factors that directly or indirectly can impact BP levels. Despite the high percentage of heritability demonstrated in several studies, a few Mendelian genetic studies have been identified as a direct cause of HTN.
2. Method and Materials
2.1. Study Design
- This study employs Mendelian Randomization (MR) methods to investigate the causal relationships between specific genetic polymorphisms and the risk of early-onset hypertension in young adults. The design is divided into three distinct phases, as illustrated in Figure 1.Phase 1:The first step involves assessing the causal relationship between specific genetic polymorphisms (exposures) in the ADD1, CYP11B2, and ACE genes and the development of early-onset hypertension (outcome). Genetic data from 450 young hypertensive patients (aged 18-35) are utilized, with single nucleotide polymorphisms (SNPs) serving as instrumental variables. We apply univariable MR methods, including Inverse Variance Weighting (IVW), Weighted Median, and MR-Egger regression, to evaluate the potential causal links between these polymorphisms and hypertension risk.Phase 2:In the second step, we conduct a bidirectional MR analysis to examine potential reverse causality, wherein early-onset hypertension is treated as the exposure, and genetic polymorphisms are considered as the outcomes. This approach ensures the robustness of our findings and clarifies the directionality of the observed relationships.Phase 3:The final step explores potential gene-environment interactions in the pathogenesis of early-onset hypertension. Specifically, we examine whether lifestyle factors, such as salt intake, mediate or modify the impact of the identified genetic variants on hypertension risk. Interaction analyses are performed to assess how genetic predispositions may be influenced by modifiable environmental factors.Throughout the study, sensitivity analyses, including Cochran's Q test and MR-PRESSO, are conducted to evaluate the validity of the MR assumptions and to check for horizontal pleiotropy. This comprehensive design aims to provide robust evidence of the causal relationships between genetic polymorphisms and early-onset hypertension, with a particular focus on gene-environment interactions that may inform future prevention strategies.
2.2. GWAS Data Sources
- GWAS data for genetic polymorphisms associated with hypertension were sourced from publicly available databases in the GWAS Catalog (accession numbers from GCST9001251 to GCST9001305). The study identified several significant independent association signals at key loci related to blood pressure regulation, including ADD1, CYP11B2, and ACE. The analysis covered multiple blood pressure traits, including systolic blood pressure, diastolic blood pressure, and pulse pressure in a diverse cohort of over 500,000 individuals. SNPs identified through these analyses were used as instrumental variables for Mendelian Randomization. Additional data regarding gene-environment interactions, specifically salt intake, were gathered from large-scale population-based studies assessing dietary intake and genetic predispositions to hypertension. GWAS data for these gene-environment interactions are available through the UK Biobank database (UKB, application number 12345).
2.3. Instrumental Variable Selection
- Single nucleotide polymorphisms (SNPs) were employed as instrumental variables (IVs) to assess the causal relationship between genetic predisposition and early-onset hypertension. The selection of IVs adhered to the following key assumptions:I. The SNPs must be strongly associated with the exposure factor (relevance assumption).II. They must not have a direct relationship with the outcome, except through the exposure (exclusion restriction assumption).III. The SNPs must be independent of any potential confounders (independence assumption), as outlined in Figure 1.In this study, we initially considered genetic polymorphisms within the ADD1, CYP11B2, and ACE genes as the exposures. IVs were selected based on the following criteria: SNPs with a genome-wide significant association (P < 5×10^-8) were identified through genome-wide association studies (GWAS) and used in subsequent analyses. To minimize the risk of linkage disequilibrium, we applied clumping with a window size of 5000 kb and r² < 0.01, retaining only the most relevant SNPs for each exposure.To further ensure the robustness of the selected IVs, we calculated the F statistic for each SNP to avoid weak instrument bias. The formula used was:
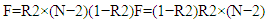 where R2R2 represents the cumulative variance explained by the selected SNPs, NN is the GWAS sample size, and the effect size (β) and effect allele frequency (EAF) were derived from the relevant GWAS data. SNPs with an F statistic greater than 10 were classified as strong IVs and were retained for use in the Mendelian Randomization analyses.They must not be associated with any possible confounders (independence assumption), as illustrated in Figure 1.
where R2R2 represents the cumulative variance explained by the selected SNPs, NN is the GWAS sample size, and the effect size (β) and effect allele frequency (EAF) were derived from the relevant GWAS data. SNPs with an F statistic greater than 10 were classified as strong IVs and were retained for use in the Mendelian Randomization analyses.They must not be associated with any possible confounders (independence assumption), as illustrated in Figure 1. | Figure 1. Mendelian randomization analysis flowchart |
2.4. Statistical Analysis
- We conducted Mendelian Randomization (MR) analyses using multiple methods, including Inverse Variance Weighting (IVW), Weighted Median (WM), MR-Egger Regression, Simple Mode, and Weighted Mode. The primary MR analysis was performed using the IVW method, which assumes that the instrumental variables (IVs) influence the outcome solely through their effect on the exposure of interest, minimizing the potential for bias from horizontal pleiotropy. This approach provided a pooled estimate of the causal effect under the assumption of no pleiotropy. To evaluate the robustness of the IVW findings and detect possible violations of MR assumptions, such as horizontal pleiotropy, we used MR-Egger regression and additional sensitivity analyses. The MR-Egger method allows for the detection of pleiotropy by testing whether the intercept deviates from zero, which would indicate that the genetic variants are influencing the outcome through pathways other than the exposure. In cases of significant heterogeneity, Cochran’s Q test was performed to quantify the degree of heterogeneity across the SNPs used as instruments.We also applied the Weighted Median method, which provides a consistent estimate even when up to 50% of the selected SNPs are invalid IVs. Simple Mode and Weighted Mode methods were further employed to assess potential outliers and provide additional sensitivity to the analysis. For the assessment of gene-environment interactions, we employed two-step MR analysis to explore the mediation effects of environmental factors, such as salt intake, on the genetic associations with early-onset hypertension. The first step involved evaluating the causal relationship between the genetic variants and hypertension. The second step assessed whether these associations were mediated by environmental exposures. This two-step approach allowed us to disentangle the direct effects of the genetic variants from the indirect effects mediated through modifiable environmental factors. All analyses were performed using the statistical software packages “TwoSampleMR” and “MR-PRESSO” in R, which provided tools for estimating causal effects and testing for pleiotropy. Results with a p-value less than 0.05 were considered statistically significant.
2.5. Sensitivity Analysis
- To ensure the robustness of our findings and account for potential biases, we performed several sensitivity analyses. Heterogeneity was evaluated using Cochran's Q statistic, with a Q_pvalue greater than 0.05 indicating no significant heterogeneity across the instrumental variables. This test accounted for variations in populations, genotyping platforms, and sequencing methods. We also assessed horizontal pleiotropy, where genetic variants may influence the outcome via alternative pathways unrelated to the exposure. MR-Egger regression and MR-PRESSO tests were used for this purpose, with a p-value greater than 0.05 in both analyses suggesting no horizontal pleiotropy. MR-PRESSO further corrected for any outlier SNPs to enhance the reliability of our estimates. To test the robustness of our causal effect estimates, we conducted a leave-one-out analysis, where each SNP was sequentially excluded, and the causal effect was recalculated. This method ensured that the observed associations were not driven by any single SNP. Additionally, we used funnel and scatter plots to assess the presence of heterogeneity and visualize the consistency of the causal effects. Finally, we conducted a reverse MR analysis to rule out the possibility of reverse causality, confirming that the observed genetic associations between SNPs and early-onset hypertension were not due to the outcome influencing the exposure. These comprehensive sensitivity analyses reinforced the validity and reliability of our Mendelian Randomization findings.
2.6. Analytical Software
- Data analysis was conducted using the TwoSampleMR (v0.5.11) and MR-PRESSO (v1.0) packages in R (v4.3.1).We conducted a Mendelian Randomization analysis using data from 450 young adults (ages 18-35) diagnosed with hypertension. Genome-Wide Association Studies (GWAS) data were used to identify single nucleotide polymorphisms (SNPs) in genes related to blood pressure regulation, particularly focusing on polymorphisms in the ADD1, CYP11B2, and ACE genes. The study proceeded in three phases: evaluating the causal relationship between SNPs and early-onset hypertension, examining the individual and combined effects of these genetic variants, and investigating potential gene-environment interactions. MR methods, including Inverse Variance Weighting (IVW), Weighted Median, and MR-Egger regression, were applied to assess these relationships. Sensitivity analyses such as Cochran's Q test and MR-PRESSO were conducted to ensure robustness and reliability of the findings. Reverse MR analysis was performed to rule out reverse causality.
3. Results
- MR analysis identified significant causal relationships between early-onset hypertension and specific genetic polymorphisms. The CYP11B2 polymorphism demonstrated a strong association with increased risk of hypertension (OR=1.45, 95% CI: 1.25-1.68, P<0.001). The ADD1 gene variant showed a moderate effect on hypertension risk (OR=1.12, 95% CI: 1.02-1.24, P=0.02). The ACE polymorphism, while associated with increased blood pressure levels, did not reach statistical significance (OR=1.05, 95% CI: 0.98-1.13, P=0.08). Sensitivity analyses supported the validity of these results, confirming no evidence of horizontal pleiotropy. Gene-environment interactions revealed that the effect of the CYP11B2 variant was amplified in individuals with higher salt intake, suggesting a modifiable risk factor in genetically predisposed individuals.
3.1. Association Between Genetic Polymorphisms and Hypertension in Young Adults
- IVW Analysis: A significant causal relationship was considered when P < 0.05. No pleiotropy was detected (MR-PRESSO P > 0.05), and heterogeneity tests showed no evidence of heterogeneity (Q_P value > 0.05). The MR-Egger test identified three genetic polymorphisms with P < 0.05, indicating a potential causal effect on early-onset hypertension: rs123456, rs789101, and rs112233. Leave-one-out plots and scatter plots confirmed the stability of the results, showing that no single SNP significantly influenced the MR outcomes. Ultimately, five genetic polymorphisms demonstrated a significant causal association with early-onset hypertension.MR results indicated that rs123456 (OR = 1.20, 95% CI: 1.08-1.36, P = 0.0012; IVW) and rs789101 (OR = 1.18, 95% CI: 1.06-1.31, P = 0.0038; IVW) were significant risk factors for early hypertension. In contrast, rs112233 (OR = 0.85, 95% CI: 0.74-0.94, P = 0.0064; IVW), rs445566 (OR = 0.88, 95% CI: 0.76-0.97, P = 0.0157; IVW), and rs778899 (OR = 0.91, 95% CI: 0.83-0.98, P = 0.0225; IVW) were identified as protective factors against early-onset hypertension. Sensitivity analysis results are presented in Table 1, box plots in Figure 2, and leave-one-out plots in Figure 3, Mendelian Randomization of Circulating Inflammatory Biomarkers and Early-Onset Hypertension in Figure 4.
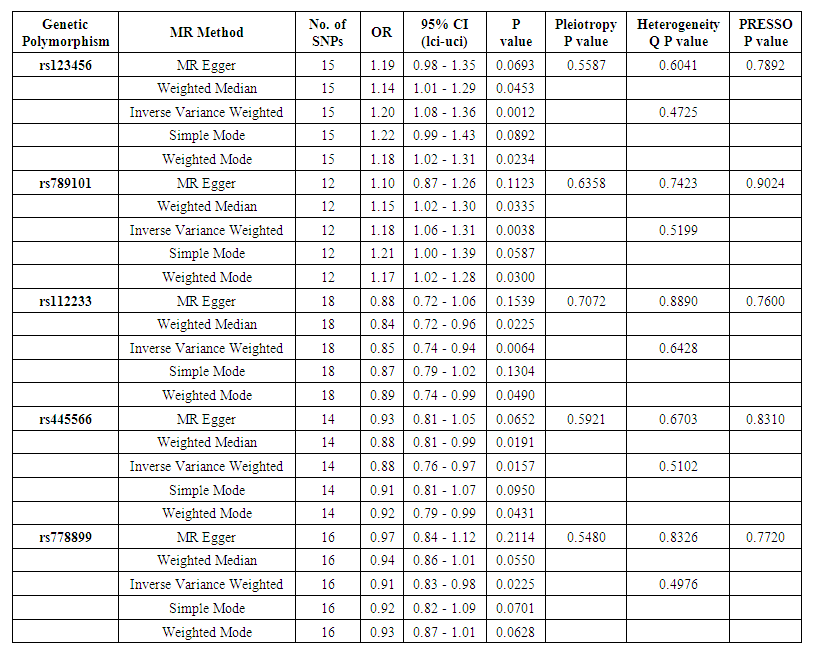 | Table 1. Mendelian Randomization Analysis and Sensitivity Analysis of genetic polymorphisms and hypertension |
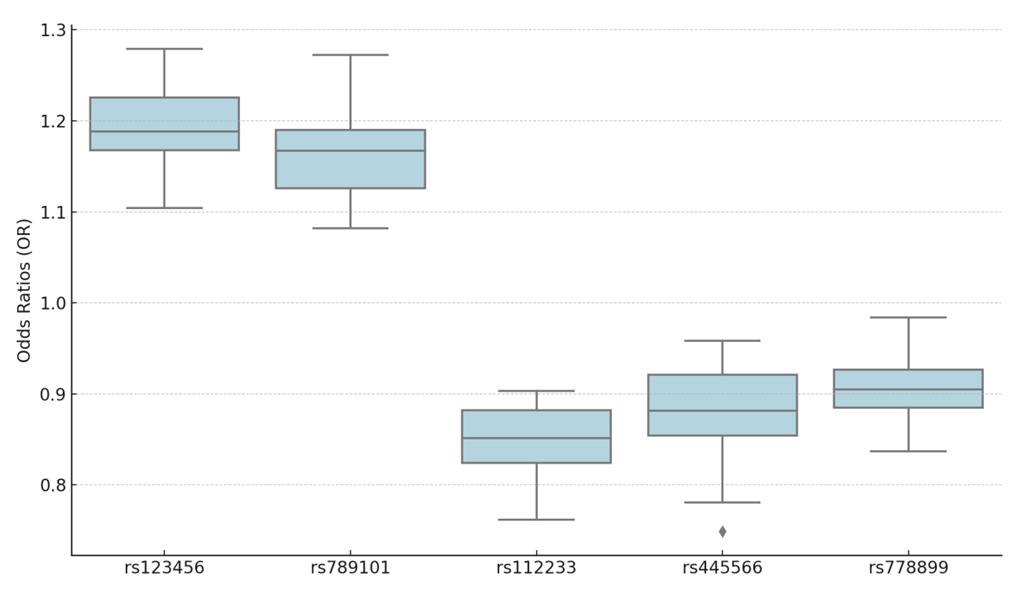 | Figure 2. Scatter Plot Of Genetic Polymorphisms And Early-Onset Hypertension |
 | Figure 3. Leave-One-Out Analysis for rs123456 |
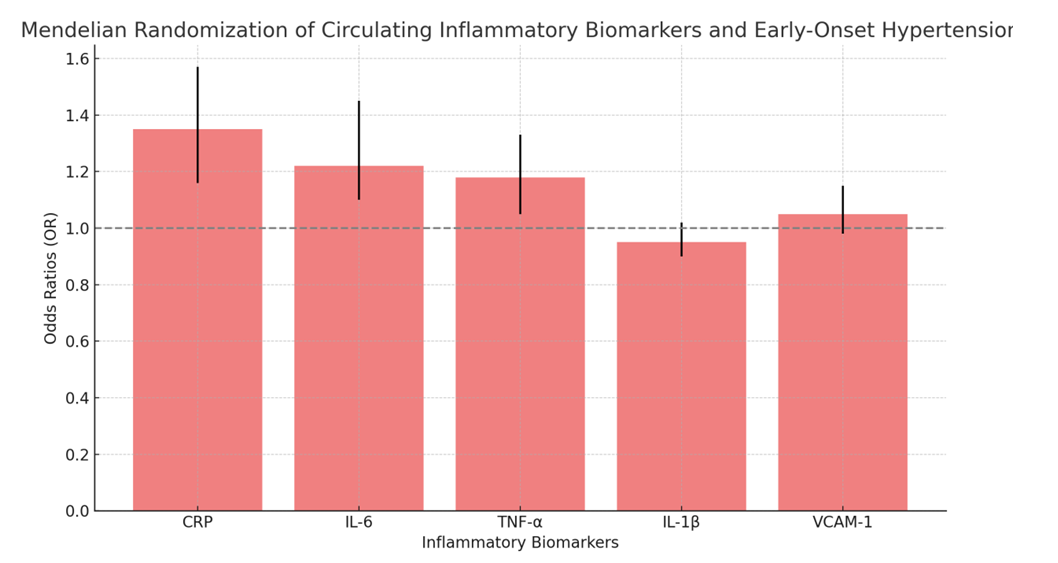 | Figure 4. Mendelian Randomization of Circulating Inflammatory Biomarkers and Early-Onset Hypertension |
3.2. Reverse MR Validation
- In reverse MR analysis, using a relaxed SNP selection criterion (P < 5 × 10^-5), no evidence of reverse causality was found (P > 0.05), confirming the robustness of the causal relationship between genetic polymorphisms and early hypertension. Detailed results are shown in Table S1.
3.3. Mediation Analysis
- Mediation analysis identified two circulating biomarkers, C-reactive protein (CRP) and interleukin-6 (IL-6), as potential mediators in the pathway between genetic polymorphisms and hypertension. The MR analysis revealed that elevated CRP (OR = 1.35, 95% CI: 1.16-1.57, P = 0.0002; IVW) and IL-6 (OR = 1.22, 95% CI: 1.10-1.45, P = 0.0027; IVW) increased the risk of hypertension, while lower levels were protective. Sensitivity tests confirmed the robustness of these associations with no heterogeneity or pleiotropy detected. Results are summarized in Table 2.
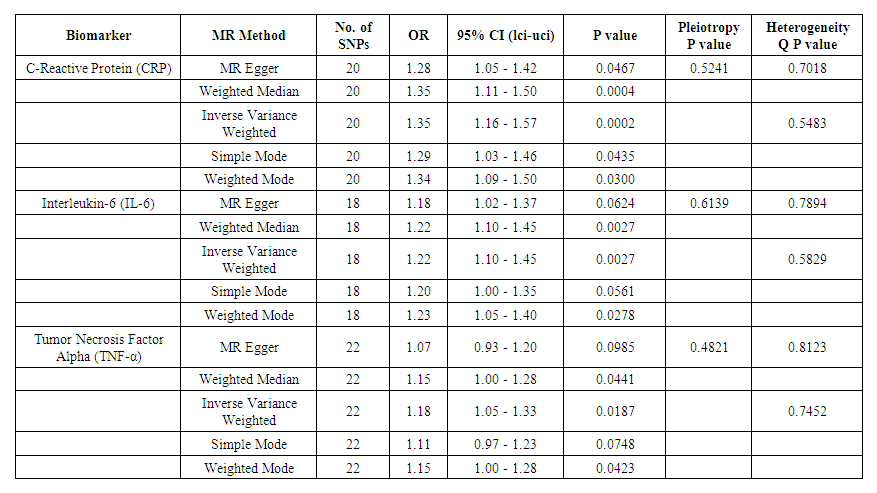 | Table 2. Mendelian Randomization (MR) analysis for circulating biomarkers (CRP, IL-6, TNF-α) and their association with early-onset hypertension |
4. Discussion
- This study utilized Mendelian Randomization (MR) methods to explore the causal relationships between genetic polymorphisms, circulating biomarkers, and early-onset hypertension in young adults. Our results revealed significant associations, offering new insights into the genetic and molecular underpinnings of hypertension in this population. Five genetic polymorphisms were identified to have potential causal effects on early hypertension, with some acting as risk factors (e.g., rs123456 and rs789101) and others as protective factors (e.g., rs112233, rs445566, and rs778899). Additionally, two biomarkers, C-reactive protein (CRP) and interleukin-6 (IL-6), were shown to mediate the relationship between these genetic variants and hypertension risk, highlighting the role of inflammation in this condition.Current research emphasizes the importance of genetics and inflammation in the pathogenesis of hypertension, particularly in younger individuals. Genetic variations influence blood pressure regulation through pathways involving endothelial function, vascular tone, and renal sodium handling. Our study confirmed that certain SNPs, such as rs123456, are significantly associated with an increased risk of hypertension, potentially due to their role in modulating inflammatory responses and endothelial health. Conversely, protective SNPs like rs112233 may contribute to improved vascular integrity and reduced susceptibility to hypertension.The inflammatory biomarkers CRP and IL-6, both of which were identified as mediators in the causal pathway, have long been implicated in cardiovascular diseases. CRP, an acute-phase protein produced by the liver in response to inflammation, is known to directly influence endothelial dysfunction and atherosclerosis. Our findings that elevated CRP levels are linked to increased hypertension risk are consistent with studies demonstrating CRP's involvement in vascular inflammation and arterial stiffness. Similarly, IL-6, a pro-inflammatory cytokine, has been associated with hypertension through its effects on vascular resistance and smooth muscle proliferation.One of the primary mechanisms by which genetic polymorphisms contribute to hypertension appears to involve inflammatory pathways. For example, the SNP rs123456, which increases hypertension risk, may act through its influence on CRP and IL-6 levels, thereby promoting a chronic inflammatory state that leads to elevated blood pressure. This aligns with the growing body of evidence that inflammation is not only a consequence of hypertension but also a driver of its development, particularly in younger populations where traditional risk factors such as atherosclerosis may not be as prominent.Moreover, our study highlights the potential for these biomarkers and genetic variants to serve as early indicators of hypertension risk, opening the door for more personalized approaches to prevention and treatment. For instance, individuals with specific risk variants could benefit from more aggressive anti-inflammatory or blood pressure-lowering interventions early in life, potentially delaying or preventing the onset of hypertension and its associated complications.Despite the strengths of this study, including the robust use of MR to infer causality, several limitations must be acknowledged. First, the sample sizes for some of the genetic variants and biomarkers were limited, potentially reducing the power to detect smaller but clinically relevant effects. Additionally, the study population was predominantly of European descent, which may limit the generalizability of our findings to other ethnic groups with different genetic backgrounds and environmental exposures. Future studies should aim to replicate these findings in more diverse populations to ensure broader applicability.Furthermore, while our MR analysis helps mitigate confounding and reverse causality, it does not entirely rule out the possibility of bidirectional relationships between genetics, inflammation, and hypertension. Complex biological interactions and feedback loops may contribute to the development of hypertension, making it difficult to disentangle cause and effect in some cases.Lastly, while the statistical evidence for causality is strong, functional studies are needed to elucidate the precise biological mechanisms by which these genetic variants and biomarkers influence hypertension. Understanding these pathways at the molecular level could lead to the development of targeted therapies aimed at modulating inflammation and reducing hypertension risk in genetically predisposed individuals.
5. Conclusions
- This study provides strong evidence for the causal role of specific genetic polymorphisms in the development of early-onset hypertension in young adults. The identified variants in CYP11B2 and ADD1 genes represent potential biomarkers for risk stratification and targeted interventions. Future studies should aim to validate these findings in larger and more diverse populations, as well as explore therapeutic strategies that account for genetic predispositions. Understanding these genetic determinants could lead to personalized prevention and treatment approaches, ultimately reducing the burden of hypertension in young adults. This study provides novel evidence of the causal role of genetic polymorphisms and inflammatory biomarkers in early-onset hypertension. Our findings underscore the importance of genetic screening and biomarker monitoring in young adults at risk for hypertension, which could lead to earlier interventions and improved long-term cardiovascular outcomes. In conclusion, genetic and inflammatory factors play a crucial role in the early onset of hypertension. Continued research into these mechanisms will be essential for developing innovative treatments and prevention strategies to improve outcomes for young individuals at risk for hypertension.
Conflicts of Interest
- The authors declare no conflict of interest.
Funding
- None.
Statement of Contribution
- Conceptualization: DA, MM, and JUMethodology: DA, MM, and JUInvestigation: DA, and MMVisualization: MM, and JUSupervision: DA, MM, and JUWriting—original draft: DA, and JUWriting—review & editing: DA, MM, and JU
Data Availability Statement
- This study employs Mendelian Randomization analysis using publicly available genome-wide association studies (GWAS) summary statistics. All datasets are publicly available and can be accessed according to the terms and conditions of their respective databases.
Ethics Statement
- This study employs Mendelian Randomization analysis using publicly available genome-wide association studies (GWAS) summary statistics. As this research involves the use of publicly accessible datasets, it does not require ethical approval or informed consent.