-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2024; 14(5): 1289-1292
doi:10.5923/j.ajmms.20241405.29
Received: Apr. 25, 2024; Accepted: May 12, 2024; Published: May 14, 2024

Features of the Clinical Course of Cystic Fibrosis in Children from Birth to Preschool Age
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAkhmedova Dilorom, Alyaviya Muzayyanakhon
Department of Hospital Pediatrics № 2, Tashkent Pediatric Medical Institute, Tashkent, Uzbekistan
Correspondence to: Alyaviya Muzayyanakhon, Department of Hospital Pediatrics № 2, Tashkent Pediatric Medical Institute, Tashkent, Uzbekistan.
| Email: |  |
Copyright © 2024 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Cystic fibrosis is an orphan (rare) hereditary genetic disease. The aim of the study – to identify the peculiarities of the clinical course of cystic fibrosis in children from birth to preschool age. Materials and Methods. Fifty-three children with cystic fibrosis aged 0-7 years were examined. Children admitted to the gastroenterology department of the Republican Specialized Scientific and Practical Medical Centre for Pediatrics of the Ministry of Health underwent standard examination stages, including a thorough history, complaints at the time of admission, clinical analysis of blood, urine and faeces, biochemical tests, and X-ray examination of the chest organs. Anthropometric measurements were performed: determination of body weight, body length/height, calculation of body mass index (BMI). Results. Children's weight and body length/height generally increase with age, but when body length/height and weight are assessed for each child individually, disharmonious development is noted. Severe degree of PEM - BAZ scores (<-3 SD) was observed in 0-1-year-old boys (62.5%) and 2 years old boys (25%), and in the other age groups of 3, 4, 6, 7 years old, mild degree of PEM (-1 to -2 SD) was determined in all boys (100%). In girls, severe PEM was detected at ages 0-1 (62.5%), 2 years (100%) and 4 years (25%), and mild PEM was diagnosed in 100% of cases at ages 3, 6 and 7 years. Conclusions. In children with cystic fibrosis, regardless of sex and age, there is a lag in physical development in all anthropometric indicators. Thus, the severe degree of PEN in boys and girls is more pronounced in the age category 0-2 years, and as they reach preschool age due to treatment, BMI indicators improve with age. Mild PEN is prevalent in both boys and girls.
Keywords: Cystic fibrosis, Children, Physical development
Cite this paper: Akhmedova Dilorom, Alyaviya Muzayyanakhon, Features of the Clinical Course of Cystic Fibrosis in Children from Birth to Preschool Age, American Journal of Medicine and Medical Sciences, Vol. 14 No. 5, 2024, pp. 1289-1292. doi: 10.5923/j.ajmms.20241405.29.
Article Outline
1. Introduction
- Cystic fibrosis is an autosomal recessively inherited disease characterized by damage to all exocrine glands, as well as the most important organs and systems [1]. Cystic fibrosis (CF) was first described in 1938 by pathologist Dorothy Andersen, who, describing cystic degeneration of the pancreas in combination with pulmonary pathology in young children, proposed the term "cystic fibrosis". Another name - "cystic fibrosis" - was introduced by S. Farber in 1944 (Latin mucus - "mucus", viscus - "viscous"), indicating the role of increased viscosity of the secretion of exocrine glands [2].Cystic fibrosis is an orphan (rare) hereditary genetic disease. Worldwide, the number of cystic fibrosis patients reaches 100,000. Given the prevalence of the disease on all continents, the greatest number of patients is observed among people of the white race. This is due to the fact that pathogenic variants of the CFTR gene are more common in Europeans. The incidence rate among Asians is 1:100000-1:350000, while among Europeans it is 1:2500-1:4500, however, it can vary even within the same country. In Russia, the incidence in newborns is 1:10000, and no gender differences have been found [3]. The disease is regarded as a pediatric pathology, but in recent decades the proportion in adults has increased by 40% [4].
2. Purpose of the Study
- To identify the peculiarities of the clinical course of cystic fibrosis in children from birth to preschool age.
3. Materials and Methods
- Fifty-three children with cystic fibrosis aged 0-7 years were examined. All children were found to have a mixed form of CF. Children admitted to the gastroenterology department of the Republican Specialized Scientific and Practical Medical Centre for Pediatrics of the Ministry of Health underwent standard examination stages, including a thorough history, complaints at the time of admission, clinical analysis of blood, urine and faeces, biochemical tests, and X-ray examination of the chest organs. Anthropometric measurements were performed: determination of body weight, body length/height, calculation of body mass index (BMI). Children's physical development was assessed according to WHO growth standards (2006) using the WHO Anthro programme (2009) using Z-score values, which are calculated in children according to age and sex. Statistical processing of the results was carried out using Statistica® version 6.0 statistical software.
4. Results
- We examined 53 children, including 31 boys and 22 girls. To assess the physical development of children with CF, anthropometric (somatometric) data on body weight, body length/height and BMI (body mass index) were calculated and presented in the table below.
|
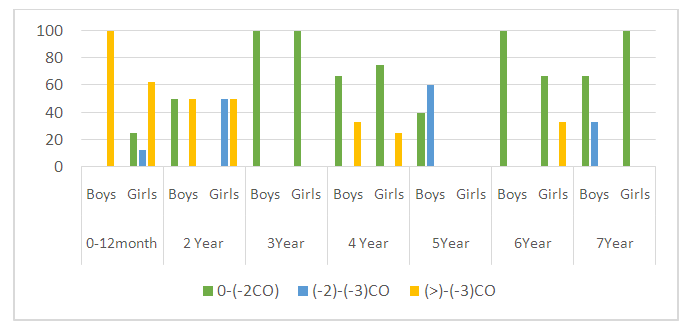 | Figure 1. Distribution of body weight Z-score value (WAZ) |
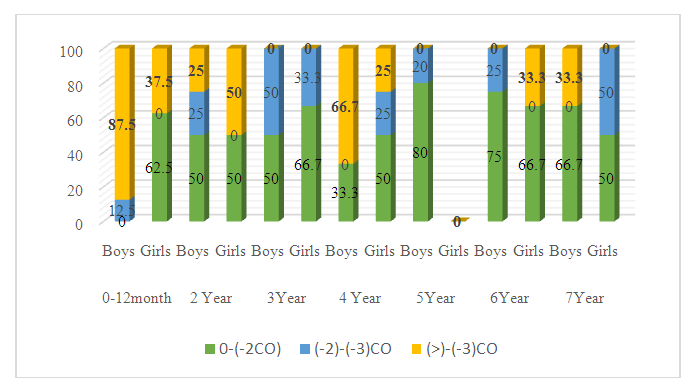 | Figure 2. Distribution of growth Z-score value (HAZ) |
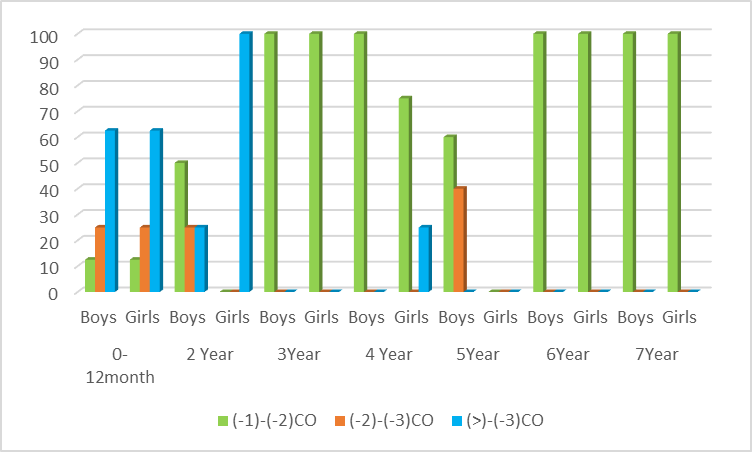 | Figure 3. Distribution of the Z-score value of body mass index (BAZ) |
5. Discussion
- In cystic fibrosis, ducts in the pancreas become obstructed with viscous secretion quite early, leading to autolysis of the gland tissue with the formation of typical fibrotic cavernous pancreatitis [5]. The development of CF symptoms is associated with the accumulation of viscous, sticky mucus in various mucin-producing organs such as lungs, sinuses, intestines, pancreas and reproductive organs [6,7]. In CF, there is an accumulation of polymeric gel-forming mucins, large O-linked glycoproteins, which are responsible for the visco-elastic properties of mucus and play a critical role in the development of this disease [8]. The clinical manifestation of cystic fibrosis of the pancreas is exocrine insufficiency, which occurs in the majority of patients and is manifested by impaired fat assimilation, steatorrhoea. In our study steatorrhea was observed in all patients.Characteristic of CF is a lag in growth and development of children, reflecting the severity of the disease, as well as the effectiveness of therapeutic measures. One of the main indicators of a healthy, physically harmoniously developed child is normal height and weight [9]. Correct measurement, analysing the results and interpreting them are the most important steps to identify growth and developmental problems in a child [10]. Decreased growth rate or weight loss is an indicator of unfavourable outcome in this disease. Many authors have found a close relationship between life expectancy and body weight [11]. According to the literature, it is important to note that the lowest anthropometric indices are noted in children with cystic fibrosis in the first year of life, which coincides with the results of our study.
6. Conclusions
- The advantage of the evaluation scale of the parametric method Z-score is that it not only allows us to accurately determine the values of all anthropometric indicators below the 3rd percentile, but also to monitor the dynamics of physical development of such children.In children with cystic fibrosis, regardless of sex and age, there is a lag in physical development in all anthropometric indicators. Thus, the severe degree of PEN in boys and girls is more pronounced in the age category 0-2 years, and as they reach preschool age due to treatment, BMI indicators improve with age. Mild PEN is prevalent in both boys and girls.Clinical manifestations of cystic fibrosis differ depending on the age of children. For example, vomiting is observed only in infants, while increased sweating and greasy stools are characteristic of CF patients of all ages.The revealed features of the clinical course of cystic fibrosis indicate that the correction of extrasecretory pancreatic insufficiency should be constantly monitored and aimed at the correction of digestion processes to physiological norm, which will improve the quality of life of patients with cystic fibrosis.
Conflict of Interest
- The authors declare no conflicts of interest or special funding for the current study.