-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2024; 14(3): 556-561
doi:10.5923/j.ajmms.20241403.04
Received: Jan. 17, 2024; Accepted: Feb. 2, 2024; Published: Mar. 2, 2024

Association of Superoxide Dismutase (SOD2) Gene Polymorphisms with Sickle Cell Anaemia (SCA) Complications in North Kordofan State, Sudan
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLMona M. S. Salama1, Mahdi H. A. Abdalla2, Nasr Eldeen Ali Mohammed Gaufri3
1Faculty of Medical Laboratory Sciences, Alneelain University, Sudan
2Department of Haematology, Faculty of Medical Laboratory Sciences, Omdurman Ahlia University, Sudan
3Department of Haematology, Faculty of Medical Laboratory Sciences, Alneelain University, Sudan
Correspondence to: Mona M. S. Salama, Faculty of Medical Laboratory Sciences, Alneelain University, Sudan.
| Email: |  |
Copyright © 2024 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Sickle cell anaemia (SCA) is an inherited blood disorder that is characterized by chronic haemolysis and episodes of many clinical complications. The number of people living with sickle cell disease globally increased from 5.46 million in 2000 to 7.74 million in 2021. This study aimed to investigate the association of superoxide dismutase (SOD2) gene polymorphisms with SCA complications. This was a case control and a hospital based study, conducted in Sickle cell aneamia center, Alkuaiti Hospital, North Kordofan state, Sudan. Following informed consent, one hundred twenty six participants were recruited to this study, 63 were SCA patients attending to Alkuaiti Hospital and 63 age and gender matched apparently healthy individuals as control group. The full blood count was done using automated hematological analyzer, Genotyping of SOD2 was determined using a PCR-RFLP method, Complications data were collected from admission and discharge records. 52.3% (n=33) from the case group were male and 47.6% (n=30) were females. There were no statistically significant differences between Hb, TWBCs and PLTs, and SOD2 genotypes p value (0.80, 0.19, 0.36) respectively. The SOD2 TT in case group was (23.8%), the CT was (49.2%), and the CC was (27%). The SOD2 TT in control group was (44.5%), the CT was (34.9%), and the CC was (20.6%). T allele frequency in case was 0.48 and in control was 0.62, while C allele frequency in case was 0.52 and in control was 0.38. This study concluded that there was a statistically significant difference in SOD2 genotypes between case group and control group with (P value= 0.04). There was no association between SOD2 gene polymorphisms and SCA complications.
Keywords: Superoxide dismutase 2, Sickle cell anaemia, Sudan
Cite this paper: Mona M. S. Salama, Mahdi H. A. Abdalla, Nasr Eldeen Ali Mohammed Gaufri, Association of Superoxide Dismutase (SOD2) Gene Polymorphisms with Sickle Cell Anaemia (SCA) Complications in North Kordofan State, Sudan, American Journal of Medicine and Medical Sciences, Vol. 14 No. 3, 2024, pp. 556-561. doi: 10.5923/j.ajmms.20241403.04.
Article Outline
1. Introduction
- Sickle cell disease (SCD) is a set of inherited hemoglobinopathies characterized via way of means of mutations that have an effect on the β-globin chain. The number of people living with sickle cell disease globally increased from 5.46 million in 2000 to 7.74 million in 2021 (Thomson et al., 2023). SCD is caused by a variant of the β-globin gene called Hb S. Hb S results from the replacement of glutamic acid by valine in the sixth position of the β-globin chain of hemoglobin (Payne et al., 2020). Inherited autosomal recessively, both copies of Hb S are required for disease expression (Ashley-Koch et al., 2000). Carrier people have one replica of the sickle variation and one replica of the normal β-globin gene (Hb AS), producing a mixture of sickle hemoglobin and normal hemoglobin. The carrier state for SCD is often referred to as a "sickle cell trait". Multiple elements decide the clinical manifestations of SCD. Both intracellular and extracellular factors influence sickling, including the types of hemoglobin in the cell and their concentrations, the level of 2,3-diphosphoglycerate (2,3-DPG), and the hydrogen ion concentration (Ellithy et al., 2015).Some of these factors are determined predominantly by genetic factors; others are environmentally modified. In addition to physiologic changes such as tissue oxygenation and pH (Ellithy et al., 2015).The complications of SCD are characterized by chronic hemolytic anemia, severe acute and chronic pain, as well as end-organ damage. SCA patients are generally well-adapted until an episode of decompensation (e.g., a severe infection) occurs (Provan et al., 2004).Severe intermittent acute pain is the most common SCD complication (Brousseau et al., 2010). Acute pain is basically associated with vaso-occlusion of sickled red blood cells. Chronic pain may be due to sensitization of the central and/or peripheral nervous system and is often diffuse with neuropathic pain features (Ballas & Darbari., 2013, Sharma & Brandow., 2020).Priapism is an undesired, persistent, and regularly painful erection that may result in erectile dysfunction.Sickle cell crisis (haematological, aplstic, vasooclusive and visceral sequestration crisis) vaso-oclusive crisis is an acute episode of pain, additionally typically known as sickle cell pain crises, or vaso-occlusive crises (VOCs). Furthermore, the frequency of VOCs, along with acute chest syndrome (ACS), is the most common predictor of death in patients with SCD (Platt et al., 1994).Bone pain affects long bones and spine, and is due to the occlusion of small vessels Triggers: infection, dehydration, alcohol, menstruation, cold and temperature changes – often no cause was found (Provan et al., 2004).Dactylitis specifically children, metacarpals, metatarsals, backs of hands and feet became swollen and tender due to small vessel occlusion and infarction (Provan et al., 2004).Acute chest syndrome is a common cause of death. Chest wall pain, sometimes with pleurisy, fever and shortness of breath. Requires prompt and vigorous treatment (Provan et al., 2004)In visceral sequestration occur sudden trapping of blood in the spleen or liver causes rapid enlargement of the organ and drop in hematocrit leading to hypovolemic shock (Nayak & Rai., 2014).Antioxidants are molecules that quench or inhibit the movement of free radicals in addition to preventing cellular damage. Antioxidants exist as enzymatic and non-enzymatic molecules within side the body (Valko et al., 2007). Enzymatic antioxidants act through metabolizing free radicals and removing from cells. Most of those antioxidant enzymes convert reactive oxidative species (ROS) to hydrogen peroxide (H2O2) and, in the end, to water (H2O), in the presence of cofactors such as manganese (Mn), iron (Fe), zinc (Zn), and copper (Cu) in a multi-step process. Major defense mechanisms against ROS include superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx) (Yoshihito., 2012).Pathological events taking place in sickle cell disease elevate free-radicals production through activation of pro-oxidant enzymes, release of free hemoglobin, and heme induced by hemolysis, which foster the Fenton reaction, modification of mitochondrial respiratory chain activity and RBCs auto-oxidation (Chirico & Pialoux., 2012, Ware et al., 2017, Schieber & Chandel., 2014).Superoxide can be formed from O2 in tissues and the occurrence of superoxide dismutase, the enzyme responsible for its elimination in all aerobic organisms (in spite of the reality that not in obligate anaerobes) indicate that the ability toxicity of oxygen is because of its conversion to superoxide (Murray et al., 2003). Superoxide dismutase protects aerobic organisms against the potential deleterious effects of superoxide (Murray et al., 2003).Three isoforms of SOD were identified. SOD1 consists of Cu and Zn within the active site (also referred to as CuZn SOD) and is mainly present in cell cytoplasm. SOD2 or Mn SOD has an active site that consists of Mn and is positioned in mitochondria. SOD3 or extracellular (EC) SOD contains Cu and Zn within the active site and is the least studied of the three SOD isoforms. SOD2 is the only antioxidant enzyme recognized to be found within the mitochondria and this has essential implications because this is a major site for the production of ROS during normal cellular metabolism (Crawford et al., 2012, Zelko et al., 2002). SOD2 gene is located on chromosome 6q25 (Pourvali et al., 2016). The SOD2 gene structure consists of five exons interrupted by four introns and the promoter, which control SOD2 expression (Wan et al., 1994). From the polymorphisms located in the coding sequence, the one called rs4880, located in exon 2. The C16T polymorphism is positioned within the mitochondrial targeting sequence and is suggested to modify the peptide structure, affecting the protein translocation and maturation into the mitochondrial matrix (Sutton et al., 2003). This substitution of C to T (GCT to GTT), that is, alanine to valine, results in structural alterations in the mitochondrial targeting domain from β-sheet to α-helix, which induces a 30-40% increase in MnSOD activity in mitochondria (Pourvali et al., 2016).This study was conducted to detect the effect of the anti-oxidant enzyme SOD2 gene polymorphisms on SCA complications.
2. Material and Methods
- This was a case control and hospital based study, conducted at the sickle cell disease center, Alkuaiti Hospital, North Kordofan State, Sudan. Following informed consent, one hundred twenty six participants recruited for this study: 63 SCA patients attending Alkuaiti Hospital, aging from 1 to 18years old, with no blood transfusion in the last three months and with no other genetic disorders. Patients under treatment affecting enzyme activities were excluded, and 63 age and gender matched apparently healthy individuals as control group. Complications data were collected from admission and discharge records, while complications were noted in a complication book and computerized system for storing data. The recorded complications are dactylitis, acute chest syndrome, painful crisis, stroke, leg ulcer, painful crisis with hepatomegaly, dactylitis with hepatomegaly, splenic sequestration, hepatic sequestration, acute heart failure, hepatosplenomegaly and heart failure with hepatomegaly. The full blood count was done using an automated hematological analyzer (Sysmex KXN 21 Japan) in Alkuaiti Hospital. Genomic DNA was extracted from blood samples (preserved at -20°C) using a DNA extraction kit (Qiagen Blood Extraction kit). Genotyping of SOD2 was examined using the PCR-RFLP method. The PCR program was initial denaturation at 95°C for 5 minutes, followed by 35 cycles of 95°C for 60 seconds, annealing at 56°C for 60 seconds and extention at 72°C for 60 seconds. The PCR was completed by a final extension cycle at 72°C for 7 minutes, the PCR product is 107bp band (Figure 1). The amplified product was digested for 24 Hours (overnight) with the NgoMIV restriction enzyme (CutSmart NEW ENGLAND Biolabs) at 37°C and electrophoresed on 2% agarose gel stained with ethidium bromide. Genotypes were determined for the polymorphism as TT (107 bp), CT (107,89,18 bp), or CC (89,18 bp) (Figure 2). Primers for SOD2 (Silig et al., 2015) (Table 1).
|

 | Figure 1. Agarose gel electrophoresis for amplified PCR product of SOD2 (107bp) |
 | Figure 2. PCR R-RFLP analysis of SOD2 gene polymorphism with NgoMIV restriction enzyme. MM : DNA ladder: 50-500bp, lanes 1,5: TT(107bp), lanes 2,3,7: CT(107,89,18), lanes4,6: CC(89,18bp) |
3. Data Analysis
- Data were analyzed using statistical package for social sciences (SPSS) version 23. In dependent T. test and One way ANOVA test were used. The correlation test used between quantitative data and study groups. Allele frequencies was estimated by counting Hardy- Weinberg equilibrium. The relations between study groups and genotypes were assessed with Chi- square (X2). The level of statistical significance was set at less than 0.05.
4. Result
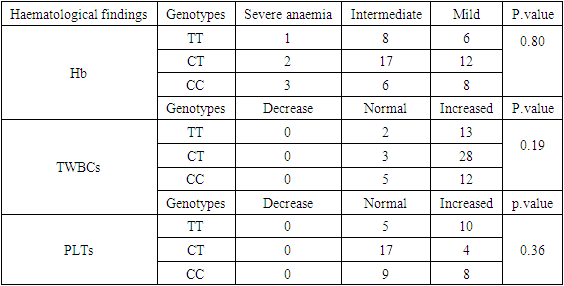
- Totally one hundred twenty six participants were recruited to this study, 50% (n= 63) was sickle cell patients consider as a case group and 50% (n=63) apparently healthy individuals as control group. 52.3% (n=33) from the case group were male and 47.7% (n=30) were females; their mean age was 8.8±5 years. The SOD2 genotypes in case group showed that the TT was (23.8%) the CT was (49.2%), and the CC was (27%), The SOD2 genotypes in control group showed that the TT was (44.5%), the CT was (34.9%), and the CC was (20.6%) (Table 2).
|

|
|

5. Discussion
- Complications of SCD are characterized by chronic hemolytic anemia, severe acute and chronic pain, as well as end-organ damage. Anaemia is chronic and patients generally well-adapted until an episode of decompensation (e.g. severe infection) occurs (Provan et al., 2004).Our study included one hundred twenty six participants recruited to this study: 63 sickle cell patients consider as a case group and 63 healthy individuals as control group, SOD2 genotypes and haematological findings of the patients were compared with control group.This study aimed to investigate the association between SOD2 gene polymorphisms and SCA complications.In this study, the common SOD2 genotype in the case group was CT (49.2%), followed by the CC (27%), then the TT (23.8%), while in the control group the common SOD2 was TT (44.5%) followed by the CT (34.9%), then the CC (20.6%), T allele frequency in the case was 0.48 and in the control was 0.62, while C allele frequency in the case was 0.52 and in the control was 0.38, with statistically significant difference between the case and control groups.Like our study, (Sogut et al., 2011) observed SOD2 genotype common frequency was CT in case group, and in control group the TT high frequency, followed by the CT, then CC. Also is in agreement with allele frequency (T allele frequency in case 0.65 and in control 0.73, while C allele frequency in case 0.35 and in control 0.27).Similar to our observation, a study by (khorsshied et al,. 2022) concluded that the commonest frequency was CT in case group, and the T allele frequency was higher in case and control than C allele frequency (T allele frequency in case was 0.55 and in control was 0.56, while C allele frequency in case 0.45 and in control 0.44), but unlike our study in control group, the commonest genotype frequency was CT, followed by TT, then CC, also there was no statistically significant difference between case and control groups.In this study there was no statistically significant differences in Hb, TWBCs and PLTs between SOD2 genotypes, our result is in agreement with (khorsshied et al,. 2022).This present study there was no statistically significant association between SOD2 genotypes and SCA complications. (khorsshied et al., 2022) reported a similar findings with no statistically significant association between SOD2 genotypes and SCD comlpications (VOC, splenic sequestration, leg ulcer) but disagreed with our study with a significant association between SOD2 genotypes and stroke.Study by Fariass et al (Fariass et al., 2018) suggest that the SOD2 polymorphism associated with low SOD activity could be a susceptibility factor for the occurrence of VOC and ASS and this disagree with our result but agree with genotype frequency of CT most common in patients.
6. Conclusions
- This study concluded that there was a statistically significant difference in SOD2 genotypes between case group and control group with (P value= 0.04). There was no association between SOD2 gene polymorphisms and SCA complications.