-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2023; 13(5): 735-739
doi:10.5923/j.ajmms.20231305.39
Received: May 12, 2023; Accepted: May 26, 2023; Published: May 27, 2023

Literature Review on the Topic: GRIN - Associated Epileptic Encephalopathy
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLShokhijakhon Shokhimardonov
Tashkent Medical Academy, Uzbekistan
Correspondence to: Shokhijakhon Shokhimardonov, Tashkent Medical Academy, Uzbekistan.
Copyright © 2023 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

GRIN-associated epileptic encephalopathies are serious genetic disorders that can cause epileptic seizures and mental retardation [1]. They are associated with mutations in genes that code for NMDA-glutamate receptors, important molecules that play a role in neurotransmission and brain plasticity. New methods of genetic diagnostics allow to identify these mutations with high accuracy and improve the diagnosis of these diseases [2]. Traditional therapies such as anticonvulsants may help reduce the frequency of epileptic seizures, but they are not effective in addressing the underlying cause [3]. Recent studies have shown that gene therapy may be a promising treatment for replacing missing or damaged genes in a patient's body, which can lead to a reduction or complete disappearance of the symptoms of the disease [1,3]. In addition, other therapies such as transcranial magnetic stimulation and deep brain stimulation can help reduce the frequency of epileptic seizures and improve the patient's quality of life [4]. In general, GRIN-associated epileptic encephalopathies are complex diseases that require a multidisciplinary approach in diagnosis and treatment. However, thanks to new methods of genetic diagnostics and molecular biology, we have new opportunities to treat these diseases and improve the lives of patients [2].
Keywords: GRIN1, GRIN2A, GRIN2D, Epileptic encephalopathy, Memantine
Cite this paper: Shokhijakhon Shokhimardonov, Literature Review on the Topic: GRIN - Associated Epileptic Encephalopathy, American Journal of Medicine and Medical Sciences, Vol. 13 No. 5, 2023, pp. 735-739. doi: 10.5923/j.ajmms.20231305.39.
Article Outline
1. Background
- GRIN-associated epileptic encephalopathies are a group of genetic disorders that cause epileptic seizures and mental retardation. These diseases are associated with mutations in genes encoding NMDA receptor subunits, which are key components of neural networks involved in learning, memory and attention [1].One of the most studied genes associated with GRIN-associated epileptic encephalopathies is GRIN2A. Mutations in this gene can cause various forms of epileptic seizures, including status convulsions, frontal lobe epilepsy, and epileptic encephalopathies [4].The symptoms of these disorders can be very varied and may include frequent seizures, psychomotor retardation, mental retardation, and other neurological problems. However, there are also many overlaps with other forms of epileptic encephalopathies, making these diseases difficult to diagnose [3,5].One of the challenges in the treatment of GRIN-associated epileptic encephalopathies is that they may be deeply rooted in genetic mechanisms. Currently, traditional treatments such as antiepileptic drugs can relieve some symptoms but often do not provide a complete cure [6].However, thanks to advances in genetic diagnosis and molecular biology, we have new options for the treatment of GRIN-associated epileptic encephalopathies. For example, gene therapy is a promising treatment that may provide the opportunity to replace missing or damaged genes in a patient's body, which can lead to a reduction or complete disappearance of the symptoms of the disease [1].Other therapies have also been investigated, including transcranial magnetic stimulation and deep brain stimulation, which may help reduce the frequency of epileptic seizures and improve the patient's quality of life [7].Despite the fact that GRIN-associated epileptic encephalopathies are rare diseases, they are of great importance for understanding the molecular mechanisms underlying epilepsy and mental retardation [8]. Understanding these mechanisms could help develop new diagnostic and treatment methods for a wide range of brain-related diseases [9].In general, GRIN-associated epileptic encephalopathies are complex diseases that require a multidisciplinary approach in diagnosis and treatment. But thanks to new methods of genetic diagnosis and molecular biology, we have new opportunities to treat these diseases and improve the lives of patients [10,11].
2. Structure and Function of the Glutamate Receptor
- All NMDAR subunits contain four semi-autonomous domains (Figure 1): amino-terminal domain (ATD), agonist-binding domain (ABD), transmembrane domain (TMD), and CTD. The dicotyledonous ABD GluN2 binds L-glutamate in the gap between the two rigid lobes S1 and S2, which form a cot, the structure undergoing pronounced conformational changes upon ligand binding [12]. The S1 lobe of the ABD, which is distal to the ion channel, forms the interface between the ABD adjacent subunits, allowing them to act as dimers. For NMDAR, one GluN1 and one GluN2 ABD form a dimer, two of which exist at each tetrameric receptor. Lobe S2, which is located proximal to the channel and contains mainly a polypeptide chain connecting two transmembrane helices (M1 and M3) through flexible linkers and a two-turn helix (pre-M1 helix) that lies parallel to the plane of the membrane [1,13]. The S2 lobe undergoes significant movement as the agonist binds to "close the cot" within the ABD of each subunit, which is an initial conformational change that may eventually lead to the opening of the ion channel pore. This combination of agonist binding and shellfish closure provides the energy to open channels in all ionotropic glutamate receptors [14].
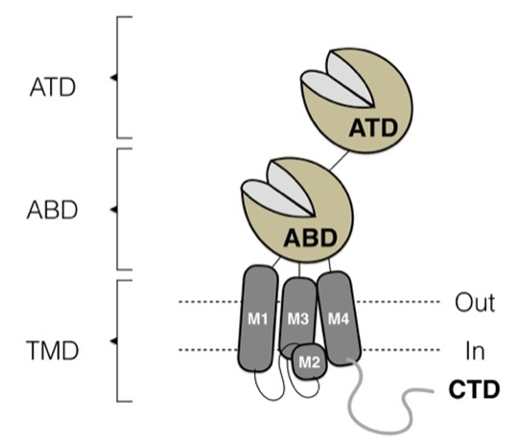 | Figure 1. NMDA receptor structure: ATD – aminoterminal domain, ABD – antagonist binding domain, TMD – transmembrane domain |
3. Diagnosis and Phenotyping
- Developmental encephalopathies are disorders associated with developmental delay, including impairments in cognition, communication, and fine motor and other motor disorders [15]. Impairments in sensory systems such as vision and hearing are also common when these developmental changes are associated with multiple modalities and are often referred to as global delay [16]. If developmental delay persists for more than 5 years, it is highly likely to lead to personality change for a long period, thereby compromising the patient's ability to perform functions necessary for independent living [15]. In such a case, the term "mental retardation" is used, which is usually classified as mild, moderate, severe or profound. Changes in developmental trajectories, including regression or loss of previously acquired skills, may be different for each disorder and are an important component of phenotyping [16,17]. Further classification of epilepsy in terms of seizure characteristics and EEG properties is a necessary feature of phenotyping [18]. Patients with GRIN variants also show altered brain structure as detected by MRI studies, aberrant muscle tone, movement disorders such as ataxia, behavioral disorders, and symptoms that can be understood from autonomic dysfunction [19].
4. GRIN Variants
- The GRIN gene family contains GRIN1, GRIN2A-D, GRIN3A-B. Of these, variants in the GRIN1, GRIN2A, GRIN2B, and GRIN2D genes have been identified in people with a variety of neurodevelopmental disorders; association of GRIN2C and GRIN3 with their association with disease remains unclear [20,21]. As expected, there are both similarities and differences between these genes in terms of the spectrum of genetic variants that appear to contribute to human pathological conditions. The GRIN genes encode different NMDAR GluN subunits. Functional NMDARs (Figure 1) require 2 subunits encoded by GRIN1 and 2 subunits encoded by either GRIN2 or GRIN3. This specificity is regulated in anatomical, developmental and neuronal subtypes, NMDAR receptors are assembled from subunits within the endoplasmic reticulum. The range of characteristics that govern assembly, such as the relative availability of any given subunit, is not fully understood. NMDARs are required for several key roles, including neuronal migration, synaptic connectivity, neuronal pruning and survival, and synaptic connectivity [22].Thus, it is not surprising that NMDAR variations play a role in human disease. GRIN1 is a phylogenetically conserved gene. Through statistical representations in a neurotypical population, GRIN1 does not tolerate both missense (Z = 6.22) and null variation (pLI = 0.98; LOEUF = 0.31). Pathogenic heterozygous missense variants have arisen de novo and are clustered next to each other in a limited number of domains with important functions such as the agonist binding domain (consisting of two different parts of the polypeptide chain), pore-forming transmembrane domains, and linker regions that connect the agonist binding domain to sometimes the channel and control the opening of the channel after the binding of the agonist (Fig. 1). The amino-terminal domain (also known as the N-terminal domain, or NTD), which is located distal to the membrane, and the C-terminal domain (CTD), which is located within the cell, are more resistant to variation and thus less likely to occur. contribute to the development of a disorder associated with GRIN1. However, this notion is subject to change with further study of the actions and binding partners for these two regions, as a full understanding of its role is still not known [23].Despite high rates of restriction, GRIN1 null variants have not yet been associated with neurological disease. Several individuals who possess heterozygous null variants of GRIN1. They are reported not to exhibit any distinct clinically significant phenotypes. One family with a homozygous GRIN1 null variant that has been associated with fatal epileptic encephalopathy has been reported, suggesting that null variants may contribute to GRIN1 associated disorders, but only when both alleles are affected. To date, this is the only report of the autosomal recessive form of any GRIN-associated disease. Despite the description of two families with rare homozygous missense variants, autosomal recessive GRIN1 related developmental disorders due to missense variation have not yet been unequivocally confirmed [23].Like GRIN1 and GRIN2B, the GRIN2D gene due to similar statistical representations in the neurotypical population missense intolerance (Z = 4.85) and zero variation (pLI = 1.00; LOEUF = 0.17). Pathogenic heterozygous missense variants usually occur de novo. GRIN2D-related disorders are the least common among GRIN disorders, so it is premature to draw conclusions about the potential clustering of pathogenic missense variants in any region of the GRIN2D-encoded protein. Null variants in the GRIN2D gene (other than large scale deletions) have not yet been reported.
5. Clinical Manifestations
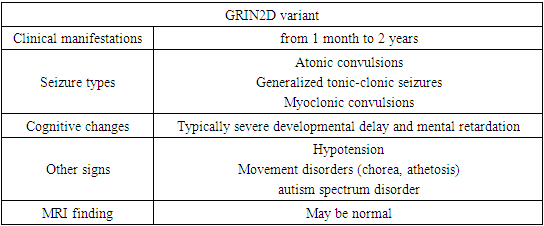
- Patients with a neurodevelopmental disorder associated with GRIN exhibit multiple disorders, including epilepsy, hypotension, and, in some individuals, movement disorders. All affected individuals assessed to date show varying levels of disability, including 5% mild, 7% moderate, 71% severe, or 17% profound disability [14,20].For convenience of perception, the main clinical signs were divided by us into groups in charts.
|
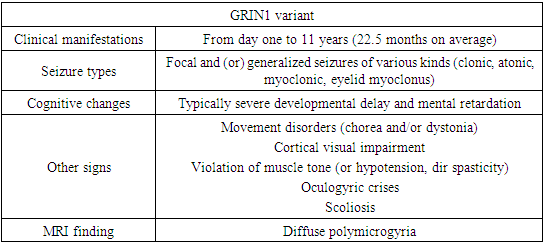
|
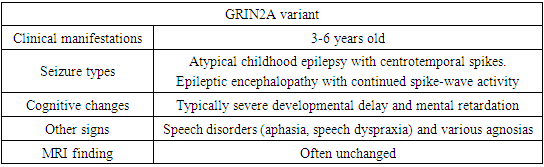
|
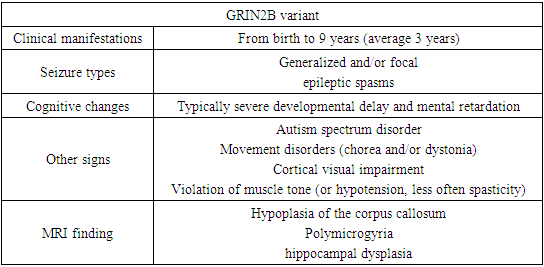
|
6. GRIN Studies of Associated Epileptic Encephalopathy
- Multiple pathogenic variants in the GRIN genes have been described as GoF or LoF for NMDAR, which gives clinicians the opportunity to potentially mitigate dysfunction through the use of either NMDAR-specific blockers/inhibitors or enhancers/potentiators. Initial evidence of principle came from treating a man with early-onset epileptic encephalopathy and drug-resistant seizures due to a pathogenic de novo GoF missense variant in GRIN2A with the FDA-approved NMDAR channel blocker memantine. This treatment resulted in a significant reduction in the frequency of seizures [5,22].Other people with GRIN variants have been treated with memantine, but only a few have been reported in the peer-reviewed literature. There is a limited number of publications, if only cases with an provably pathogenic GRIN variant with a confirmed effect of GoF on NMDAR are considered. For GRIN1, a recent GoF missense variant that has shown increased efficacy for an FDA-approved set. NMDAR channel blockers have been described, which creates opportunities where clinical treatment preferentially blocks variants but not wild-type receptors. The affected individual responded positively to memantine with a significant reduction in seizure frequency and severity of spasms, with evidence of efficacy in seizures/spasms worsened during accidental discontinuation of memantine [24]. For GRIN2A, only one patient is reported in the literature to have a significant reduction in seizure frequency after addition of memantine followed by dextromethorphan, similarly. Four patients have been reported for GRIN2B [25]. All of these individuals demonstrated subtle and subjective improvements in claw function, behavior, or sleep quality. However, none had quantifiable improvements in these modalities to objective assessment [26]. For GRIN2D, two patients have been reported who demonstrated a mild to moderate improvement in seizure frequency after the addition of memantine to the treatment regimen. In only one patient with the LoF GRIN variant, subjective behavioral improvements, as well as improved sleep and motor development, were reported with L-serine administration [24]. In these studies, a safety assessment cannot be provided due to the small number of individuals studied; it may be dangerous to generalize these case studies further. These early but limited studies suggest that targeted therapies for GRINs are possible, but there remains an urgent need to evaluate these treatments in well-designed, double-blind, placebo-controlled clinical trials that systematically and quantify several parameters of therapy safety and efficacy [23].