-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2021; 11(7): 512-514
doi:10.5923/j.ajmms.20211107.02
Received: May 30, 2021; Accepted: Jun. 21, 2021; Published: Jul. 15, 2021

Pathomorphology of Dilyatation Cardiomyopathy
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLOripova Ozoda Olimovna, Isroilov Rajabboy Isroilovich
Samarkand State Medical Institute "Department of Pathological Anatomy", Uzbekistan
Correspondence to: Isroilov Rajabboy Isroilovich, Samarkand State Medical Institute "Department of Pathological Anatomy", Uzbekistan.
| Email: |  |
Copyright © 2021 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

The aim was to study the pathomorphological changes in myocardial structures in DCM. Materials were obtained from autopsy DCM cases in RPAC over the last 20 years (2010-2020) and the autopsy protocol and medical history were analyzed. Sudden development of heart failure in the right ventricle and left ventricle in DCM, cardialgia and angina pectoris; cardiac arrhythmias, often with ventricular fibrillation, ventricular extrasystoles, and conduction block, have been observed in some cases with thromboembolism. Histologically, the following types of pathomorphological changes in the myocardium and endocardium were observed in DCM, ie excessive growth of interstitial connective tissue, development of myxamatosis, lipomatosis in some places. The main changes characteristic of DCM were dilatation of muscle fibers, ie thinning of muscle fibers, thinning, fragmentation and homogenization of cardiomyocytes, disordered placement of nuclei and deformation and dystrophy due to changes in the environment.
Keywords: Heart, Cardiomyopathy, Dilated cardiomyopathy, Myocardium, Morphology, Microscopy
Cite this paper: Oripova Ozoda Olimovna, Isroilov Rajabboy Isroilovich, Pathomorphology of Dilyatation Cardiomyopathy, American Journal of Medicine and Medical Sciences, Vol. 11 No. 7, 2021, pp. 512-514. doi: 10.5923/j.ajmms.20211107.02.
1. Introduction
- Cardiomyopathy is a primary lesion of the myocardium of the heart, characterized by inflammation, tumors, specific cardiomegaly unrelated to ischemia, exacerbated heart failure, and arrhythmias [1,2]. Therefore, it is called an idiopathic disease of the myocardium, ie of unknown origin, based on the development of dystrophic and sclerotic changes in cardiomyocytes. Therefore, cardiomyopathies are always impaired ventricular function. Secondary cardiomyopathies resulting from primary disease are myocardial damage that develops in hypertension, vasculitis, symptomatic arterial hypertension, autoimmune diseases of connective tissue, myocarditis, myocardial dystrophy and other pathological conditions. The following types of primary cardiomyopathy are distinguished: dilated, hypertrophic, restrictive, and arrhythmogenic.In addition, depending on the occurrence of cardiomyopathies in certain geographical regions, morphological changes in the myocardium, the name may change, for example: African cardiomyopathy, which is more common on the African continent; stagnant cardiomyopathy, in which the heart cavities dilate and the blood stagnates; constrictive cardiomyopathy - disruption of the wall of the heart cavity due to subendocardial fibrosis; obliterated cardiomyopathy - a decrease in the volume of the heart cavities due to the appearance of thrombi attached to the valves; familial cardiomyopathy is an autosomal-dominant type of disease that occurs in members of a family, develops at the age of 10-20 years, manifested by fainting, shortness of breath, pulsation in the chest, arrhythmias [2,3,4].Of these, dilated cardiomyopathy (DCM) is the most common type, characterized by significant enlargement of the heart cavities, hypertrophy of the myocardium, and decreased contractility. These symptoms start at 30-35 years of age. This disease is extremely common, the incidence rate reaches 2500: 1, leading to the highest mortality as heart failure. The incidence rate in women and men is 1: 5. Idiopathic DCM usually develops at a young age [5,6].The causes of primary DCM have not been studied to date. Possible causes may be: Infectious diseases: Coxsackie, herpes, influenza, caronovirus and other viral infections, hereditary factors, DCM develops from the addition of an autoimmune process after myocarditis. Toxic effects: most common under the influence of alcohol; drug effects: anthracyclines, doxorubicin; effects of heavy metals: cobalt, mercury, arsenic, lead. It can be caused by systemic autoimmune diseases of the connective tissue. Incomplete metabolites of tyrosine-tryptophan produced in pheochromocytoma tumor may result. Neuromuscular diseases, i.e. Duchenne-Becker and Emery-Dreyfus muscular dystrophic diseases. It can also be caused by metabolic disorders, i.e. metabolic, endocrine, mitochondrial diseases, selenium and carnitine deficiency. Idiopathic DCM occurs in 20–35% of cases, depending on more than 20 genes and loci. It usually passes in an autosomal dominant way, sometimes linked to the X chromosome. It was found that in DCM, too, hypertrophy and mutated genes in CM are damaged (a - actin, a - troponins). Cases of hypertrophic CM becoming DCM have also been reported.The purpose of the study is study of pathomorphological changes in myocardial structures in DCM.
2. Methods
- Over the last 20 years (2010-2020), autopsy DCM cases have been obtained in RPAC. The autopsy report and medical history were analyzed. Autopsity material was re-examined and histological preparations were re-examined from cardiac fragments. Clinical-anamnestic analysis showed that in our material DCM is clinically manifested by the following symptoms: heart failure develops suddenly in the right ventricle and left ventricle; cardialgia and angina pectoris; cardiac arrhythmias, often with ventricular fibrillation, ventricular extrasystole, and conduction block, have been demonstrated in some cases with thromboembolism. The clinic of the disease was found to develop in a nonspecific manner, and sudden death of patients was observed. Clinical examinations revealed an increase in blood pressure, first in the left ventricle and then in the right ventricle. Often with left ventricular failure: shortness of breath, bruising, with asthma and lung tumors, followed by right ventricular failure: acrocyanosis, pain and enlargement of the liver, ascites, edema, swelling of the jugular veins, heart pain, persistent pain with nitroglycerin . On objective examination of the patient revealed chest deformity, dilated left and upward cardiomegaly, suffocation of the tones at the apex of the heart, systolic murmur, gallop rhythm. Often, severe forms of hypotension and arrhythmia, such as paroxysmal tachycardia, extrasystole, mercatilly arrhythmia, and blockages, have been identified in dilated cardiomyopathy. Electrocardiography showed left ventricular hypertrophy, conduction disturbances, and arrhythmias. Exo CG revealed diffuse myocardial injury, sudden dilation of the ventricles of the heart, dysfunction of the left ventricular dystonia. The main criterion for DCM disease was a 45% decrease in blood flow by the left ventricle, and it was confirmed that the ventricular cavity was reduced by 6 cm in diastole.
3. Results
- Morphologically, DCM is manifested by eccentric hypertrophy and dilatation of the heart cavities. In general, the left side of the heart was found to be damaged, only in some cases, i.e., in 1.7%, was the right ventricle altered. Histologically diffusely revealed the presence of interstitial sclerosis spreading to cardiomyocytes, hydropic dystrophy of cardiomyocytes. Cardiomyocyte atrophy was observed in 50% of cases.The results of histological examination of cardiac tissue in cases of DCM showed the presence of the following types of pathomorphological changes in the myocardium and endocardium. The main morphological changes were observed in the interstitial tissue, ie excessive growth of connective tissue, the development of myxamatosis, lipomatosis in some places. Similar morphological changes were found in the vascular wall, i.e., sclerosis developed from the proliferation of pericytes, and the basal membrane and elastic fibers were mixed with myxamatosis. If we study and analyze the changes in the interstitial connective tissue, we can see that the interstitial substance is prone to swelling and myxamatosis. In addition, the appearance of lymphoid cells in the interstitial tissue, the proliferation of macrophages, that is, the development of morphological changes specific to the autoimmune process (Fig. 1). Interstitial connective tissue cells are found to develop changes in various forms, such as metaplasia and dysplasia. Most importantly, the transformation of connective tissue histioblasts and histiocytes into fat cells is determined. It is observed that the interstitial connective tissue is strongly swollen, both fibers and cells are in disarray, the cytoplasm of some histioblast cells is enlarged and vacuolated due to the formation of fat (Fig. 2). As a result, there is an increase in adipose tissue in the myocardial interstitial tissue and the appearance of infiltration of lymphoid cells around it (Fig. 3).
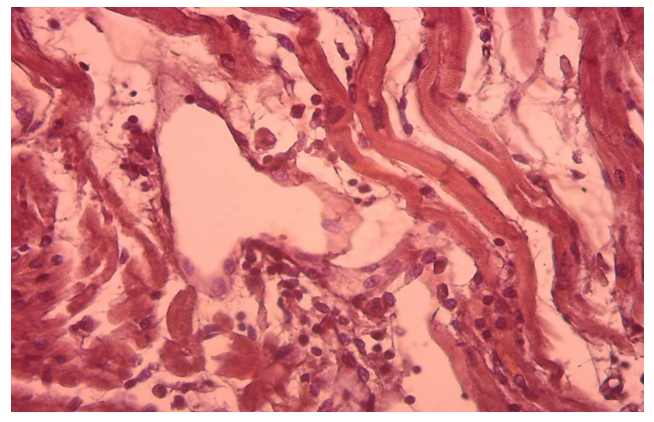 | Figure 1. Interstitial tissue tumor, myxomatosis, lymphoid cell formation, and development of autoimmune process. Paint: G-E. X: white |
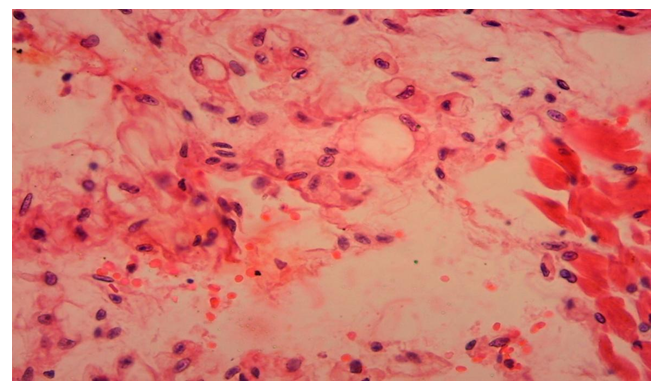 | Figure 2. Metaplasia of connective tissue cells into fat cells in myocardial interstitial tissue. Paint: G-E. X: white |
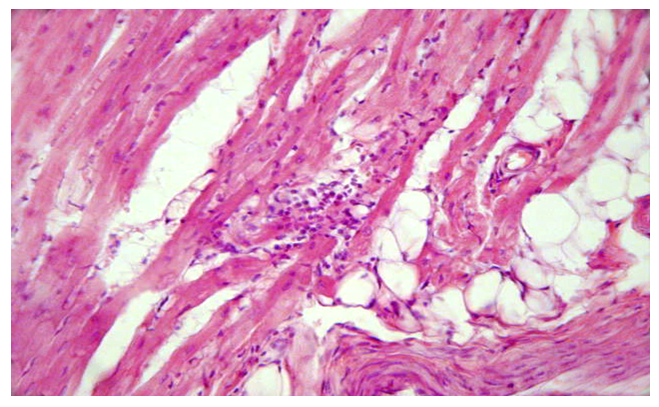 | Figure 3. Occurrence of adipose tissue and lymphoid infiltration in myocardial interstitial tissue. Paint: G-E. X: white |
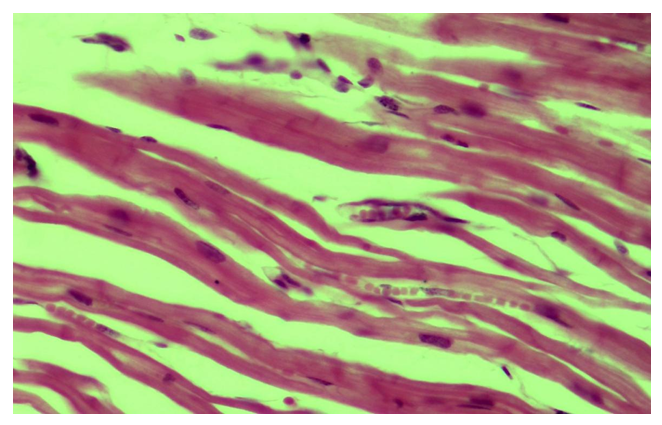 | Figure 4. Rupture of myocardial muscle fibers, loss of transverse ligaments, homogenization and myolysis. Paint: G-E. X: white |
4. Conclusions
- 1. Heart failure in DCM develops suddenly in the right ventricle and left ventricle; cardialgia and angina pectoris; cardiac arrhythmias, often with ventricular fibrillation, ventricular extrasystoles, and conduction block, have been observed in some cases with thromboembolism.2. Histologically, the following types of pathomorphological changes in the myocardium and endocardium were observed in DCM, ie excessive growth of interstitial connective tissue, the development of myxomatosis, lipomatosis in some places.3. Most importantly, the transformation of connective tissue histioblasts and histiocytes into fat cells, i.e., the cytoplasm of some histioblast cells expands and vacuoles due to the formation of fat, resulting in the growth of adipose tissue in the myocardial interstitial tissue and infiltration of lymphoid cells around it.4. The main changes characteristic of DCM were dilatation and elongation of muscle fibers, ie thinning of muscle fibers, thinning, fragmentation and homogenization of cardiomyocytes, disorganization of nuclei and deformation and dystrophy due to changes in the environment.