-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2019; 9(0): 384-388
doi:10.5923/j.ajmms.20190910.06

Clinical-age Characterization of Pheochromocytomas in the Gender Aspect
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAlimoukhamedova Gulrukh Aibekovna, Khalimova Zamira Yusufovna
Ya. Kh. Turakulov Tertiary Center for the Scientific and Clinical Study of Endocrinology, Uzbekistan Public Health Ministry, Tashkent
Correspondence to: Alimoukhamedova Gulrukh Aibekovna, Ya. Kh. Turakulov Tertiary Center for the Scientific and Clinical Study of Endocrinology, Uzbekistan Public Health Ministry, Tashkent.
| Email: |  |
Copyright © 2019 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

The work was initiated to study clinical implications in patients with pheochromocytomas. Materials and methods: Clinical observations among 51 patients (23 men, 45.1%; 28 women, 54.9% and 6 children, 11.8%) with pheochromocytomas undergoing both outpatient and inpatient treatment at the tertiary Center for the Scientific and Clinical Study of Endocrinology, Uzbekistan Public Health Ministry, are at the core of the study. The patients’ age ranged from 13 to 66 years, mean age was 38.7 ± 13.7 years. General clinical, biochemical, hormonal and instrumental methods of investigation constitute the algorithm of the study. Results and discussion: Our study on age-related peculiarities of patients with pheochromocytomas demonstrated presence of pheochromocytomas in all age groups, but predominantly (54.9%; χ2=39.2; р<0.0001) in the young patients (18-44 years). The right localization was found in most patients (62.7%; χ2=5.65; р=0.02), in 13 (25.5%) and 6 (11.8%) the left and bilateral localization was found. Clinical manifestations of pheochromocytoma were highly variable. High arterial pressure observed in all patients (n=51, 100%) was the main symptom of the diversified PHEO clinical picture. Mean age at the first elevation of arterial pressure was 35.4 ± 13.4 years; in 40 patients (78.4%; χ2=30.7; р<0.0001) taking place at the age under 45, that is at the working age, while occurring after 45 in 11 (21.6%). Conclusions: Most pheochromocytomas (54.9%) presented in young people with insignificant predominance of women as compared to men (54.9% versus 45.1%, respectively). Arterial hypertension was registered in 100% of patients being the main clinical symptom of the disease. Arterial hypertension was the mixed one; that is with arterial pressure elevation > 200 mm Hg in the setting of the initially increased arterial pressure. In 15 patients (29.4%) arterial hypertension was constant; sharp rise in arterial pressure in the setting of normal arterial pressure could be seen in 5 (9.8%).
Keywords: Pheochromocytoma, Adrenals, Arterial hypertension, Adrenalectomy
Cite this paper: Alimoukhamedova Gulrukh Aibekovna, Khalimova Zamira Yusufovna, Clinical-age Characterization of Pheochromocytomas in the Gender Aspect, American Journal of Medicine and Medical Sciences, Vol. 9 No. 0, 2019, pp. 384-388. doi: 10.5923/j.ajmms.20190910.06.
- Pheochromocytoma is a hormonally active tumor from the chromaffin tissue producing catecholamines and clinically manifesting with arterial hypertension syndrome of various manifestation rates, as well as with multiple metabolic disorders [1]. The prevalence of PPGLs, that is, of pheochromocytomas (PHEOs) and extra-adrenal paragangliomas (PGLs) is estimated ranging between 1:65000 and 1: 2500, the incidence in the USA varied from 550 to 1600 cases per year [2]. The 4th and 5th decades of life are the most frequent period for the tumors to onset, more frequently in women than in men (55.2% versus 44.8%) [3,4]. A pathological production and secretion of adrenalin, noradrenalin and dopamine resulting in the pronounced lesions of organs and systems is responsible for multiplicity of the tumors’ clinical manifestations.The catecholamine-producing tumors typically present with the diverse and variable manifestations suggestive of many diseases that is why pheochromocytoma is frequently called “great mimic” [2]. Persistently high arterial pressure resistant to a drug therapy and cardiovascular symptoms are a central sign of pheochromocytoma [1]. The incidence of the tumor is estimated ranging from 0.005% to 0.1% in the general population and from 0/1% to 0/2% in the adult population with arterial hypertension. According to some reports, the diagnosis is delayed in up to 70% of cases after many years of life with the uncontrolled or untreated arterial hypertension or posthumously [5]. The uncontrolled (untreated) hormonal activity of pheochromocytoma may have fatal consequences [6]. In a series of patients with PHEOs found posthumously, 75% died of myocardial infarction or the stroke [7]. As the risk of lethal outcome or severe complications is increased, early diagnosis and prompt treatment of the “time bomb” are crucial [8]. Pheochromocytoma and its complications are a cause of disablement and not infrequently deaths of patients of working age [9]; timely diagnosis and adequate treatment are a priority in current clinical medicine. The work was initiated to study peculiarities of clinical manifestations in patients with pheochromocytomas.
1. Materials and Methods
- Clinical observations among 51 patients (23 men, 45.1%; 28 women, 54.9% and 6 children, 11.8%) with pheochromocytomas undergoing both outpatient and inpatient treatment at the tertiary Center for the Scientific and Clinical Study of Endocrinology, Uzbekistan Public Health Ministry, are at the core of the study. The patients’ age ranged from 13 to 66 years, mean age was 38.7 ± 13.7 years.General clinical, biochemical, hormonal and instrumental methods of investigation constitute the algorithm of the study. Clinical picture being evaluated, arterial hypertension, its severity, character and duration of the disease were in the focus of the study. The general clinical investigation constituted thorough collection of complaints, medical and life history, assessment of somatic and endocrine statuses, complete clinical examination with arterial pressure measured and body mass index calculated, as well as complete blood count and urine analysis. The biochemical investigation included measurements of the serum potassium, sodium, chlorine, calcium, lipid profile, the fasting glucose and the 2h postprandial glucose, as well as in some cases the oral glucose tolerance test, measurements of the glycated hemoglobin, coagulogram, creatinine, urea and glomerular filtration rate. To study hormonal profile, plasma metanephrines and normetanephrines, plasma aldosterone and aldosterone-to-renin ratio, ACTH and cortisol were measured. The adrenal MSCT with the contrast enhancement in some cases was used as a specific instrumental method. The ECG and ophthalmoscopy were mandatory.To evaluate the peculiarities of clinical manifestations, the mean and standard deviations (M ± SD), as well as the frequency of occurrence was used. Correspondence of numerical data to normal law of distribution was assessed. The differences between mean values of independent and dependent samples were assessed by one-factor ANOVA analysis. χ2 criterion was used to analyze significance of differences between qualitative characters. Significance for all analyses was set up at p<0.05.
2. Results and Discussion
- The patients were distributed by age in compliance with the WHO classification (Table 1) to study age peculiarities.
|
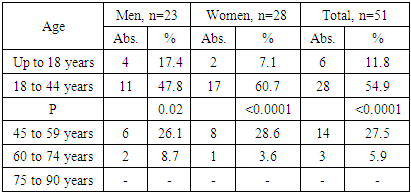
 | Figure 1. Pheochromocytoma of right adrenal in a 26-year old woman: a round solid mass with sharp contours (size 4.1 x 3.6 x 4.5 cm, density +43+45 HU) |
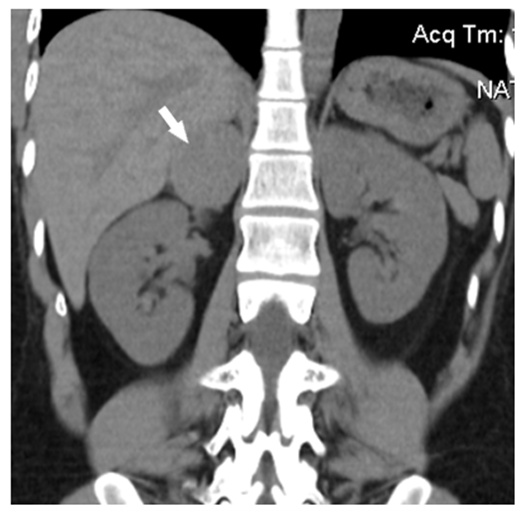
 | Figure 2. Pheochromocytoma of right adrenal in a 47-year old man: a round homogenous mass with sharp regular contours can be seen emerging from the medial limb of adrenal gland (density +43+47 HU, diameter 2.6 cm) |
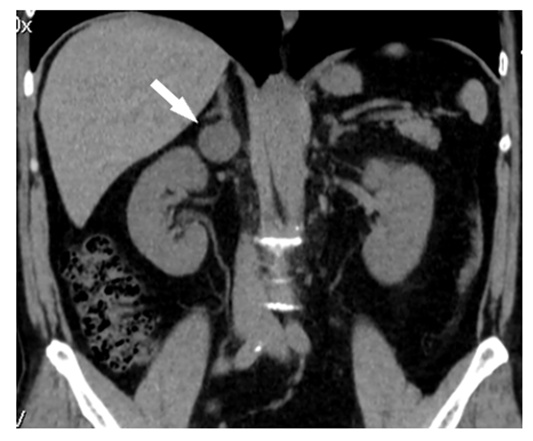
 | Figure 3. Hematoxylin and eosin staining of alveolar variant of pheochromocytoma. Multiple alveoli with inner surface consisting of large epithelial, round and polygonal cells with vacuolated cytoplasm can be seen under the thick connective tissue capsule. Nuclei with moderate hyperchromatism and atypism are situated eccentrically. The layers of hyalinized tissue can be seen between alveoli. Foci of gelatinization, as well as an interstitial edema are evident. The cortex is with extensive hemorrhaging foci and decline of lipids in the cytoplasm of adenocytes |
3. Conclusions
- 1. Most pheochromocytomas (54.9%) presented in young people with insignificant predominance of women as compared to men (54.9% versus 45.1%, respectively). 2. Arterial hypertension was registered in 100% of patients being the main clinical symptom of the disease. Arterial hypertension was the mixed one in 60.8%, persistent in 29.4% and with sharp rises in blood pressure in 9.8%. Arterial hypertension was found concomitant to vegetative-visceral disorders, such as headaches (96.1%), palpitations (74.5%), sweating (67.6%), fever (58.5%), nervousness (56.9%), tremor during hypertensive crisis (51%) and vertigo (49%).3. Familial medical history is of additional concern, since cardiovascular diseases in relatives of men under 55 years were found in 43.1%, and in relatives of women under 65 years of age in 31.4%; in particular, hypertensive disease (58.8%), stroke (19,6%), IHD (13.7%), hypertensive crisis (7.8%), myocardial infarction (7.8%), in addition to malignancies (35.3%), diabetes mellitus (15.7%), kidney disease (5.9%) and pheochromocytoma (3.9%). Familial disease increases risk of pheochromocytoma in a patient with an adrenal mass. 4. The right localization was found in most patients (62.7%); in 25.5% and 11.8% the left and bilateral localization were respectively found. On MSCT scans pheochromocytomas were rounded, oval or irregular with sharp or indistinct contours, homogenous or heterogeneous structure, sometimes with anechogenic areas corresponding to the zones of destruction. The sizes of tumors varied from 1.8 to 11.6cm. Analysis of denstitometric characteristics demonstrated that the tumors had high density in the range from 27.9±11.7 to 39.8±14.1 HU.