-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2016; 6(1): 16-22
doi:10.5923/j.ajmms.20160601.03

Stroke in Sickle Cell Disease, Risk Factors Comparative Study
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLElan Hadi Hasan1, Talib Abduljaleel Jasim2
1Al-Qadissyia University, College of Medicine, Iraq
2Al-Kufa University, College of Medicine, Iraq
Correspondence to: Elan Hadi Hasan, Al-Qadissyia University, College of Medicine, Iraq.
| Email: |  |
Copyright © 2016 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Background silent and overt stroke comprise 10-20% of complication of sickle cell disease. There was great risk of morbidity and mortality, some time with neurocognitive dysfunctions. Objective: identify the most important risk factor for neurologic events using MRI and TCD as aid for diagnosis. Method: descriptive study carried out over one year period in Al-Zahraa hospital thirty patients with sickle cell disease enrolled. All tests done as part of follow up, which include serum ferritin, blood count, MRI, and TCD. Results of the study: from total 30 patients enrolled, only one patient found to have overt stroke, two other with silent stroke and 14 patient got abnormal TCD study. Most of CNS events found in patients with low hemoglobin (p value 0.015), low red blood counts (p value 0.001), and those with high hemoglobin S concentration. Blood transfusion noticed to be protective against stroke. Conclusions: risk of cerebral stroke was related to, severity of anemia, high concentration of hemoglobin S. Recommendations: regular monitoring of cerebral blood flow by TCD and MRI decreased morbidity and mortality, together with improvement in social, and conditional affairs of patients.
Keywords: Sickle cell, Stroke, Trans cranial Doppler
Cite this paper: Elan Hadi Hasan, Talib Abduljaleel Jasim, Stroke in Sickle Cell Disease, Risk Factors Comparative Study, American Journal of Medicine and Medical Sciences, Vol. 6 No. 1, 2016, pp. 16-22. doi: 10.5923/j.ajmms.20160601.03.
Article Outline
1. Introduction
- Sickle cell disease (SCD) refers to a group of disorders in which the red blood cell undergoes sickling when deoxygenated. Sickle red cell adhesion to vascular endothelium appears to be the nidus for vaso-occlusion, rather than simply stiff sickled cells blocking the microvasculature. If a starting point can be identified, it might be the large numbers of stress reticulocytes which have adhesion molecules expressed on their surfaces that are not present on more mature red cells [1, 2]. Typical manifestations include recurrent pain and progressive incremental infarction [5]. Children with sickle cell disease, a chronic hemolytic anemia, present with a wide variety of neurological syndromes, including ischemic and hemorrhagic stroke [3, 4].Concentration of Sickle hemoglobin (Hb S) is a factor in that gelation of HbS occurs at concentrations greater than 20.8 g/dL (the normal cellular Hb concentration is 30 g/dL). Fetal hemoglobin (HbF) has an inhibitory effect on gelation [5].Sickle RBC adhesion in post capillary venules can cause increased micro vascular transit times and initiate vaso-occlusion [6].Stroke may be defined as an acute syndrome caused by restriction of cerebral blood flow with resultant ischemia and neurologic symptoms transient ischemic attack may be considered as a brief episode of neurologic dysfunction caused by focal brain ischemia, with clinical symptoms typically lasting less than one hour, and without subsequent neurologic or neuroradiographic evidence of cerebral infarction [7]. A “silent” cerebral infarct has been defined as an area of abnormally increased signal on intermediate and/or T2 weighted pulse MRI (magnetic resonance imaging) sequences compatible with brain infarction, without associated neurologic symptoms with a normal neurologic examination [9-11].Overt stroke and silent stroke occurs in SCD as 10% and 20% respectively [8]. computed tomography (CT) scan and MRI imaging remains the initial test of choice for assessment and trans cranial Doppler (TCD) is the imaging of choice for stroke prevention [9]. In patients who have presented with acute stroke, large infarcts in an arterial territory or in the border zones between anterior and middle cerebral arteries have been described [10] in association with endothelial hyperplasia, fibroblastic reaction, hyalinization and fragmentation of the internal elastic lamina, and thrombi in large and small vessels. In addition to the typical small necrotic lesions in the border between the cortex and the subcortical white matter, acute demyelination [11]. The wide spread use of MRI and MR angiography (MRA) in the 1990s resulted in an appreciation of the extent and location of lesions not only associated with overt stroke but also those related to silent cerebral infarcts. Effective stroke prevention has become possible through the utilization of trans cranial Doppler ultrasonography (TCD) [12]. TCD is a safe, noninvasive, well tolerated, relatively low-cost procedure in which the velocity of blood flow can be measured in intracranial vessels using an ultrasound probe placed over the temporal bone to screen for cerebrovascular disease [13]. In a comparison with conventional angiography, TCD showed a sensitivity of 90% and specificity of 100% for the diagnosis of abnormality. Use of Doppler ultrasonography had become an important way to identify extra cranial carotid stenosis based on a derivation of the Bernoulli principle, which is that in the area where the vessel is narrowed, the velocity of blood flowing in that vessel is increased. TCD seemed a more direct diagnostic approach. The TCD was also attractive because it was harmless, painless, and generally well tolerated, even by children, and did not require sedation. It was also portable and low cost compared with other methods. It was known that cerebral blood flow and flow velocity as estimated by TCD are increased in the anemic condition owing to the lowered oxygen-carrying capacity of the blood. It was evident from work by Adams et al [14] that the evolving velocity standards of TCD for stenosis in adults would not apply in SCD owing to the impact of young age and anemia. The early use of TCD in adults had shown that the normal velocity (time-averaged mean of maximum blood flow as opposed to systolic or diastolic) was approximately 60 cm/s. It was determined that the expected middle cerebral artery velocity in healthy children was approximately 90 cm/s, and in children with SCD without overt stroke it was approximately 130 cm/s, elevated on the basis of young age and severe anemia. When the angiogram showed severe stenosis, the velocity was always greater than 190 cm/s and often much higher except in cases of severe stenosis, in which very low velocities were recorded (70 cm/s) [15]. Arbitrary velocity cut off points less than 170 cm/s represented “normal” or average risk, 170 to 199 cm/s was called “conditional” and was associated with moderate risk, and 200 cm/s or greater was called “abnormal” or high risk. A risk of stroke during the next 36 months of 13% per year was observed. The use of chronic transfusion to prevent secondary stroke, which can occur in 60%-90% after the primary stroke, has been widespread since the 1970s and has reduced the risk of this occurrence to approximately 15%, and the discontinuation of chronic transfusion after 1–2 years, or even 5–12 years (16), is associated with a high risk of stroke recurrence.
2. Aim of the Study
- To identify the common risk factor for cerebrovascular events in sickle cell disease using neuroimaging and Trans cranial Doppler aids.
3. Patient and Methods
- A retrospective analytic study carried out on 30 patients with SCD who has been diagnosed previously on the bases of history, examination and Hb Electrophoresis (Variant 1, Biorad 1999) registered in the hereditary blood diseases center in Al-Zahraa Teaching hospital in AL-Najaf, from the first of February to the end of November 2013, inclusion criteria includes patients have SCD with HbS more than 50% of total Hb, both sex, age range 2 to 16 years. Exclusion criteria include sickle cell trait, patient more than 16 years and patients who have other diseases like congenital heart diseases.Full history include, age of diagnosis the disease, family history of SCD, transfusions, chelating agent, Hydroxyurea, (dose and compliance) physical examination include any abnormal neurological finding included one sided weaknesses and / or sensory changes (numbness, tingling sensation), balance, vision loss, slurring of speech, facial asymmetry, headache, seizures, and behavior disturbance.Sample of blood taken (as part of usual patient’s follow-up) with EDTA tube and sent for CBC. (hematology analyzer, Ruby 2010), and serum tube sent for serum ferritin by Mini Vidas, Biomeuirx 2009). Imaging studies carried out using brain MRI and TCD, brain MRI done in the department of radiology in Al-Zahraa hospital, (Open Magnet field 0.2 tesla, Siemens 2004), the reports was revised by the radiologist of the unite. TCD done in Al-Forat Al-Aosat center for the neurological diseases in Al-Sadir hospital in Najaf, (Ez-Dop 1071/ DWL Company) and the report revised by the specialist physician in the unit.Statistical analysis was done using SPSS program V.20, data were expressed and comparisons of proportions was performed using independent T- test, P-value of < 0.05 was considered as statistically significant. Logistic regression analysis was also done for the analysis of different variables, and for each variable the mean, standard deviation and 95% confidence interval (CI) were assessed by this method.
4. Results
- Total number of patients included are thirty, 16 males and 14 females, age range 2-16 years (mean 11.06±4.16) from the total only one patient have overt stroke, occurred 3 years ago as sudden onset of loss of consciousness followed by gradual improvement, other patients have normal history and examination. Most of them have good school performance but some of them stop there education due to low socioeconomic state.Diagram 1 demonstrate, that one (3.33%) patient show feature of old insult by MRI (large encephalomalacia area seen at left tempero-parietooccipital region with flecks of calcification), 2 (6.66%) patient have features of silent stroke (2 lacunar infarctions in the left periventricular area and the other show 1 small lacunar infarction in the right periventricular area), 27 (90%) patients have normal MRI.
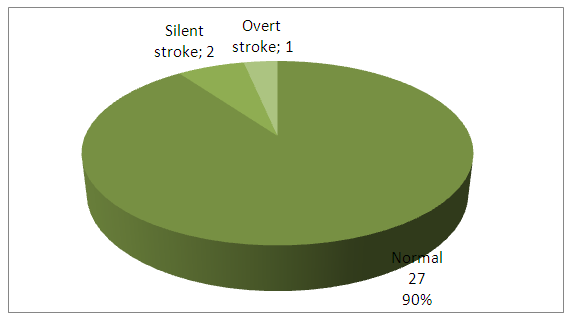 | Diagram 1. MRI finding for studied patients |
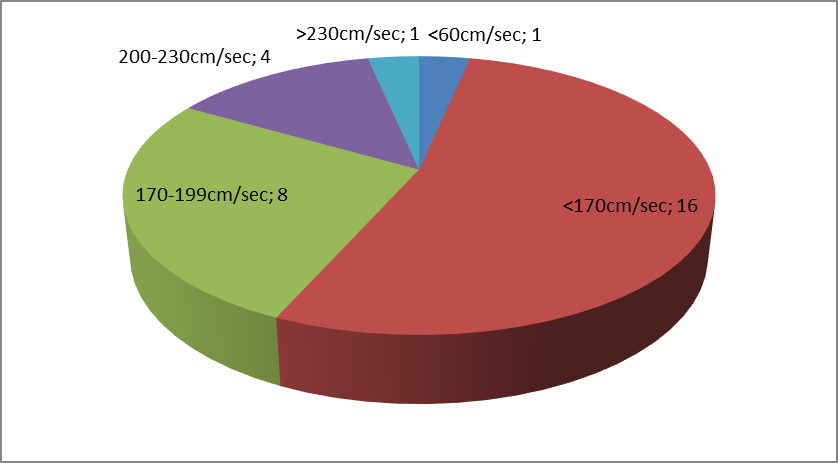 | Diagram 2. Speed of blood velocity at MCA using TCD |
- TCD finding as following, (diagram 2) 16 patients (53.33%) have cerebral artery blood velocity of MCA less than 170 cm/sec., while 8 (26.66%) have velocity of MCA 170-199 SC/sec., 4 (13.33%) have velocity of MCA 200-230 cm/sec., one (3.33%) have velocity of MCA more than 230 cm/sec., and one (3.33%) have velocity of MCA less than 50.
5. Patients Categorized in to Two Groups
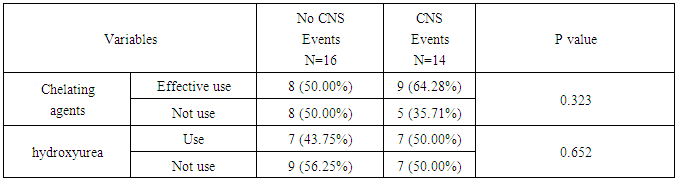
- First group with CNS events include 14 patients (46.66%), this group includes those with overt and silent stroke and patients who are at risk for stroke that appear as abnormal blood velocity at middle cerebral artery(MCA) by TCD. second group were sixteen patients (53.33%), without any neurological finding. For the age difference there was no statistical difference between both groups since 10 patients (62.50%) out of 16 who have no CNS events are above 10 years and 10 patients (71.42%) out of 14 who have CNS events are above 10 years.Regarding the hematological variability, table 1 demonstrate that from 16 patients without CNS events only one patient (6.25%) have low hemoglobin level, while 11(78.57%) out of 14 patients with abnormal neurologic events demonstrate low hemoglobin level, p value is significant (P=0.015). This finding strengthened by finding that, patients who received more than 180 milliliter of packed red cell per year had less CNS complications than those who were on episodic transfusions, the p-value was less than 0.05. Regarding R.B.C count 9 patients (56.25%) have low R.B.C count out of 16 patients with no CNS events while low count seen in 12 patients (85.71%) out 14 with CNS events, p value was 0.001. regarding white cell count and plate late there was no statistical difference between both groups.
|
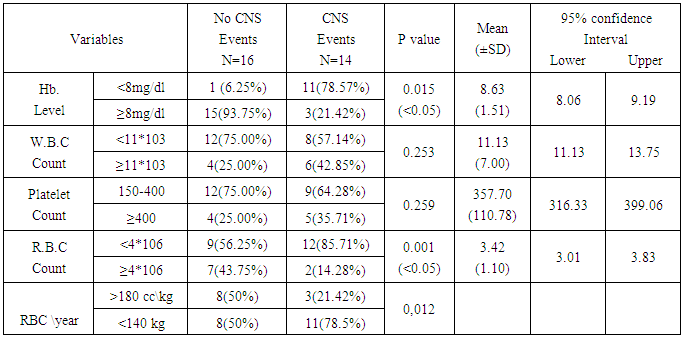
|
|
6. Discussion
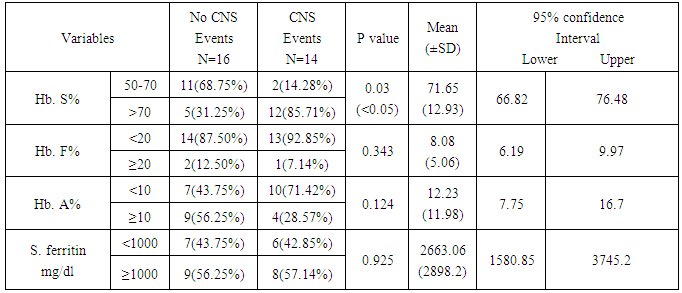
- As a complication, overt stroke and silent stroke can occur in 10%, 20% respectively in CSD [17]. The silent stroke associated with neurocognitive dysfunction, diminished educational performance, and is a risk factor for additional silent strokes or overt stroke but the best management of silent cerebral infarcts is unknown at this time [18] so it is much better to focus on the prevention of these complications rather than treating it. This study aimed to clarify the most important hematological risk factors for neurological complications in our patients using MRI, and TCD. For the effect of hemoglobin level this study found that 11 (78.57%) out of 14 patient with neurological events show low level of Hb. In comparison with only 1 (6.66%) out of 16 patient with out, neurological events, p value (P=0.015) this mean that neurological events associated with lower Hb. level. The same result done in a large prospective multicenter study in France, (Bernaudin et al., 2000) [19].Eight patients (50%) out of 16 patients without any neurological events group received more than 180 milliliter of packed red cell per year trying to elevate their hemoglobin level more than 8 g/dl, while only 3 (21.42%) out of 14 patient with neurological events blood transfusions in the same way in the last 2 years, p-value(P=0.012). This can explained by some patients live far away from the city, and some have poor economic state to comply well with the their visit to the hospital, also Low hemoglobin, high white blood cell, seem to be risk factors for overt ischemia in SCD. For hemorrhagic stroke, only low hemoglobin and high white blood cell count were found to be predictors in sickle cell CNS disease. Real data indicate that nocturnal hypoxia, related to anemia, might increase the risk of CNS events [20]. American study have shown that children with initially abnormal TCD velocities (>200cm\s) treated with regular transfusion for 30 months or more, resulted in reduction of flow to less than 170cm. Discontinuation of transfusion program led to an unacceptably high rate of TCD reversion back to high risk(>200cm\s) velocity [21]. The long term effect of transfusion, including iron over load has recently become easier to manage with introduction of an oral iron chelator. Transfusion also has significant effect in reduction of inflammatory response and prevents reduction of nitrous oxide level through cessation of hemolysis in SCD [22].It was found that blood transfusion associated with lower ischemic and lower cerebral blood velocities. The use of chronic transfusion for the prevention of primary stroke is recommended in the late 1990s [23].Regarding the platelet count, unlike foreign studies, there was no statistical difference between the two groups, in general platelet count show mild increment in SCD [24], but there is an evidence that the high platelet count involved in the mechanism of stroke and lead to neurological events [25], This may be due to small number of the studied patients comparing with these studies.White blood cell count show no difference between the two groups however leukocytosis occur in SCD [24] but leukocyte increases to higher levels in hemorrhagic stroke rather than ischemic stroke [24]. Platelets and leucocyte activation, which might affect endothelial function, are inversely related to the mean overnight oxygen saturation in child with SCD, and there is evidence for increased level of markers of cellular and endothelial adhesions [26]. Regarding R.B.C count, 12 (85.71%) out of 14 patients of neurological events show decrease RBC count compare with 9 (56.25%) out of 16 patients without neurological events this make a significant difference between the two groups (P=0.001). Reduction of red blood cell count reflects the degree of anemia due to chronic hemolysis, with its pathological and noxious effect on microcirculation of the brain [27] Hemoglobin-S- level measurement by Hb electrophoresis reflect the severity of the disease and then presence of complications [24] ,12 (85%) out of 14 patients of neurological events group show HbS level more than 70% of total Hb, while 5 (31.25%) out of 16 patients who have no neurological events got this level (P=0.030) this mean that the neurological complications is more with the increasing HbS level.Trying to decreased the level of hemoglobin S level to below 30%, using either Hydroxyurea or blood transfusions, will help to decreased the risk of stroke recurrence to between 10% and 20% [28].HbF has a role of a protective agent against HbS by the prevention of polymerization of HbS under deoxygenating state thus decrease complications. Many factors lead to increase its level, the genetic presentation of the disease itself (sickle-thalassemia or HbS-B thalassemia) or by using drugs that lead to increase its level like Hydroxyurea [25]. The level of HbF should be more than 20% in order to predict its effect on the disease. Since all studied patients got fetal hemoglobin below 20% there was no statistical difference this may be due to most of them have HbS level more than 70%, and all patients were not on Hydroxyurea therapy.Serum ferritin level increases in SCD as results of chronic hemolysis or due to frequent blood transfusion [29] there is no statistical significant difference between the two groups, this may be due to its direct effects on the CNS is doubtfully since it cannot cross blood brain barrier. There are some evidence that support worsening clinical out come in the presence of sever iron over load. Serum ferritin maybe unreliable for estimating body iron because vaso-occlusive crises are associated with elevated serum ferritin. However monitoring of serum ferritin may be useful while the patients in the steady state [30].It has been found that young children with SCD were at high risk of stroke [15].The highest mortality occurs at ages below 10 years and above 50 year, (58, 61) the age of the patients also affects the presence of the CNS complications as the highest mortality occurs in ages below 10 years and above 50 years [25]. For age as risk factor this study denote no significant deference between those with, neurological abnormalities and those without changes, this due in part to that most of our patient were bellow ten years of age and they were on regular transfusion program rather than episodic transfusion, trying to keep their hemoglobin at reasonable level. The use of Hydroxyurea decrease CNS events by increasing HbF level but in our study there was no statistical difference between the two groups, which may be explained by the fact that most of our patients were not on effective dose, due to un availability of the drug and its expensive price in the market. It has been known that Hydroxyurea have appositive effect on serum nitric oxide level which was reduced in sickle cell patients as a result of presence of free hemoglobin in the circulation. This positive effect might improve the circulation due to modifying the vasodilation of circulation [31].There are emerging data that Hydroxyurea may have beneficial effect in primary or secondary prevention of stroke in children, its role relative to that of blood transfusion has been defined by prospective trials. Until more data available blood transfusion is the modality of choice in treatment and prevention of stroke in SCD [32].
7. Conclusions
- 1- Risk of CNS events (overt or silent stroke) is directly related to low hemoglobin level, high percentage of sickled hemoglobin and low or irregular blood transfusion.2- Chelating agents and Hydroxyurea, although improving the general condition in patient with CSD, they were not able to prevents or decrease the consequences of CNS complications.
8. Recommendations
- 1- Early detection of factors that increased risk of stroke is important to raise our awareness about the CNS complications using frequent TCD and MRI especially for those aged less than 10 years.2- Maintenance of hemoglobin level by frequent blood transfusion with iron chelating agent to the level that improve brain perfusion and prevents cerebral arteries stenosis.