Mohamed Siddig Mohamed Ali1, Mahmood Haj Ali Salih Qalalweh2
1Department of Pathology, College of Medicine, King Faisal University, Sadi Arabia
2Director of Laboratory, Al Razy Hospital, Jenin, Palestine
Correspondence to: Mohamed Siddig Mohamed Ali, Department of Pathology, College of Medicine, King Faisal University, Sadi Arabia.
| Email: |  |
Copyright © 2014 Scientific & Academic Publishing. All Rights Reserved.
Abstract
The study intended to determine the frequency of B-Thalassaemia and other abnormal hemoglobins among the major ten Palestinean families using blood samples collected from 263 unrelated individuals belonging to those families. Samples were selected randomly proportional to the size of each family and subjected for Complete Blood Count (CBC), Gel electrophoresis and Chromatographic separation of HbA2. Results showed that elevation of hemoglobin F (Hb-F) was close to that of hemoglobin A2 (Hb-A2) and clearly observed in Aboalrob and Torokman followed by Ziud, Daraghmeh, Jaradat, Amarneh and Qlalweh. Elevation of the combination (Hb-F & A2) was significantly associated with microcytosis (low MCV) and hypochromia (low MCHC). Haemoglobin S (Hb-S) was highly detected in Qalalweh (41%) followed by Noairat (27%), Aboalrob (14%), Zaqzooq (9%) and Amarneh and Zekarneh (5%). Hb-S was significantly associated with low MCH and normal MCV and MCHC. On the other hand, the comparison between Hb Electrophoresis and Ion-exchange Resin laboratory methods, which used for the detection of abnormal hemoglobins revealed significant increase of the detection limit of Hb Electrophoresis than Ion-exchange Resin. The study concluded that the frequency of B Thalassaemia is quietly different among Palestinean families in addition to the presence of Hb-S. Therefore, it was strongly recommended establish a social protocol for marriage accompanied with medical investigation and supervision to reduce this risk. Also, the study recommend to apply Hb Electrophoresis since it is more reliable.
Keywords:
Beta Thalassaemia Prevalence Jenin Palestine
Cite this paper: Mohamed Siddig Mohamed Ali, Mahmood Haj Ali Salih Qalalweh, Prevalence of Beta Thalassaemia in Jenin Area in Palestine, American Journal of Medicine and Medical Sciences, Vol. 4 No. 5, 2014, pp. 180-185. doi: 10.5923/j.ajmms.20140405.08.
1. Introduction
Thalassaemias are group of genetic disorders of haemoglobin synthesis, result from reduced rate of production of one or more of the globin chains of haemoglobin. They are divided into α, β, γ, δβ or γδβ thalassaemias, according to which globin chain is produced in reduced amount. Beta thalassaemias are the most important types of thalassaemia because they are so common and produce severe anaemia [1] whether affects all haemoglobin produced (homozygous), or only half of the haemoglobin produced (heterozygous) [2].The clinical syndromes associated with Thalassaemia arise from the combined consequences of inadequate haemoglobin production and of unbalanced accumulation of one type of globin chain, which causes Hypochromic Microcytic anaemia and leading to ineffective erythropoiesis and haemolysis [3]. In homozygous beta thalassaemia, deficiency of Beta chain synthesis results in an accumulation of alpha chains. Because of their great instability, free alpha chains aggregate to form insoluble inclusions in bone marrow erythroid precursors [4] which interfere with red cell maturation and their passage through the microcirculation, particularly in the spleen hence, prematurely destroyed [1]. The anaemia acts as a stimulus to erythropoietin production and this cause expansion of the bone marrow which lead to serious deformities of the skull and long bones therefore, the spleen is being constantly bombarded with abnormal red cell [1].There is no definitive treatment except marrow transplantation, and in countries where the disease is common, a major strategies effort for prevention [5]. Bone marrow transplantation and stem cell transplantation are therapeutic modalities that are also available for the severe thalassemic patient with a success rate of 50% - 80%, although a rigorous risk is suspected [6]. The symptomatic management of severe β-thalassaemia includes regular blood transfusion, splenectomy if hypersplenism develops, and the administration of chelating agents to manage the problem of iron overload that preiptated by multible blood transfusions [5].About 3 % of the world's population (150 millions) people carry Beta Thalassaemia genes. The highest prevalence is found in Sardinia ranging from 11 to 34 % followed by Greece, where there are an estimated 3500 (5 to 15 %) individuals with Thalassaemia major [4]. In South East Asia, the disease occurs in a line starting in Southern China and stretching down through Thailand and the Malay Peninsula through Indonesia to some of the Pacific Island populations. In this region and in some of the Mediterranean Islands, gene frequencies are ranging between 2% and 30% [1]. Screening policy exists on both sides of the Island of Cyprus to reduce the incidences of thalassaemia, since the programs implantation in the 1970s, has reduced the number of children born with the hereditary blood disease from 1 out of every 158 births to almost zero [7]. In Jenin area, in the northern part of Palestine, where 200.000 persons live, there are about 150 children having the disease (Thalassaemia major) according to hospitals’ records. Those patients live in poor areas with limited resources hence, the only effective treatment (bone marrow transplantation) is unaffordable, so they are subjected for regular blood transfusions. Better availability of blood and an increase in the number of transfusion centers will led to a higher life expectancy of thalassaemia patients as they would live up to the age of 25 to 30 years. However, many communicable diseases could in principle be transmitted by blood and this account for a rather formidable list of illnesses ranging from allergy to venereal diseases [8]. Therfore, it is important to screen persons in endemic areas such as Jenin for this abnormality to know the carrier states, upon which many precautions could be taken to reduce the incidence of the disease thus, such complications and fatalities could be avoided. The study also may help in detecting other congenital abnormalities associated with Thalassaemia, which could be avoided among the next generations. Therefore, objectives of the study are:• To find out the distribution of Beta Thalassaemia and other Hb variants in individuals belong to 10 different families in jenin area, Palestine. • To find out the relationship between abnormal Hb variants and red cell indices, white cell count, platelets count and gender.• To compare between Hb electrophoresis and Ion-exchange Resin laboratory methods which used for the detection of abnormal hemoglobins.
2. Materials and Methods
The design of the study is descriptive cros-sectional. It was conducted in Jenin area which is located in the northern part of Palestine during the period from Jan 2006 to Dec 2007. All the major Plastenian tribes (10) were recruited for the present study; however, 263 unrelated individuals were selected proportional to the size of the family. Blood samples (2ml) were collected by venipuncture from all individuals in seperated vaccutainer tube containing 2.4mg K2-EDTA anticoagulant [7]. Blood samples were analysed after a proper labeling and collection for Complete Blood Count, Gel electrophoresis and chromatographic separation for HbA2 quantitation. Complete Blood count (CBC) was performed by fully automated cell counter (Medonic CA 620-Balder version 4 from Boule Medical / Stockholm-Sweden) as per manufacturer instructions.Gel electrophoresis was performed using both kit and machine of Sebia System, France made. Hydragel Hemoglobin (E), K20, Ref. 3010 was used. Sebia system complete (HRYS-2) was used.The hydragel Hemoglobin K 20 was designed for separation of the normal Hemoglobins (A and A2), and for the detection of the major hemoglobin variants: S, D, C, or E by electrophoresis on alkaline agarose gel. The resulting electrophoregrams were evaluated by the use of densitometry and giving concentrations of individual fractions.The assay was performed on hemolyzed and washed RBCs. The hemoglobins are separated by electrophoresis on alkaline gels and the fractions are quantitated after being stained with Amidoblack. For quantitation of HbA2, an anionic exchange resin for the separation of HbA2 in the samples was used. The sample was hemolyzed and passed through an anionic exchange resin and HbA2 in the elute is then specifically eluted in strictly controlled conditions of pH and ionic strength and quantified by direct photometric reading at 415 nm. Percentage of HbA2 = Absorbance of HbA2 / Absorbance of the total Hb × 30.5 (30.5 is the concentration of standard) [7].
3. Results
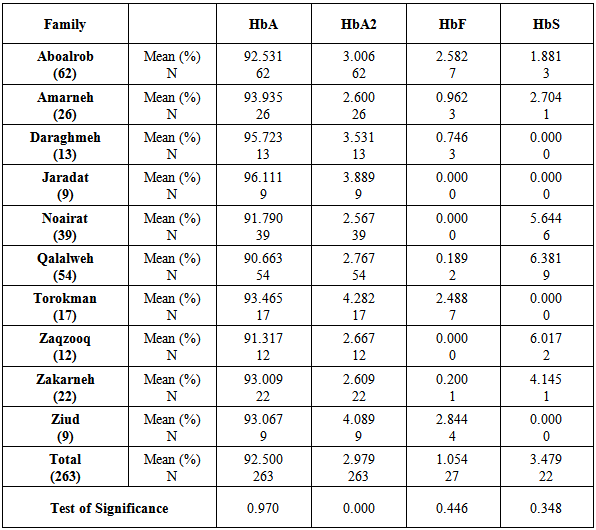
Table 1 shows significant variation of HbA2 in the studied families, and the similarity of the concentrations of HbA, HbF and HbS in the same families.Table 1. Means of HbA, HbA2, HbF, and HbS concentrations among the major palestinean families
 |
| |
|
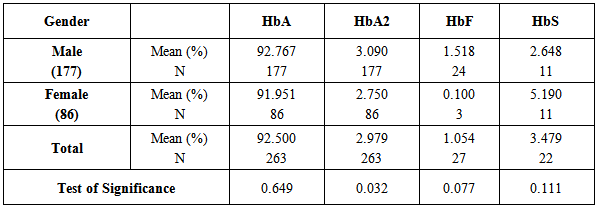
Table 2 indicates the significant increase of HbA2 in males rather than females While, HbA, HbF, and HbS are egually distributed in males and females.Table 2. Means of HbA, HbA2, HbF, and HbS concentrations in relation to gender in the major palestinean families
 |
| |
|
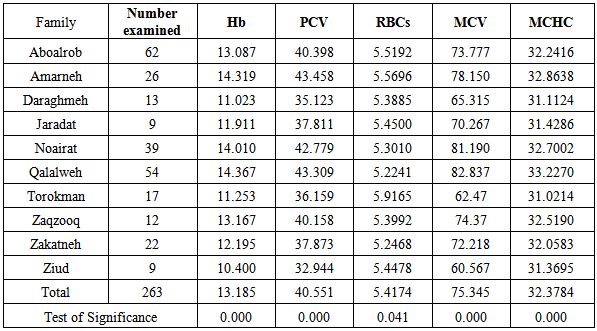
As illustrated in table 3, total Hb concentration, PCV, RBCs, MCV and MCHC values are significantly variable in the different major tribes in Palestine.Table 3. Means of the total haemoglobin (Hb), Packed Cell Volume (PCV), Red Blood Cells (RBCs), Mean Cell Volume (MCV), and Mean Cell Haemoglobin Concentration (MCHC) among the major tribes in Palestine
 |
| |
|
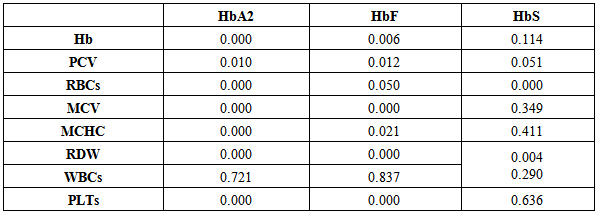
Table 4 illustrates the significant decrease of Hb, PCV, and RBCs count in individuals with HbA2 and HbF.Table 4. Test of significance (P-value) between different types of haemoglobins and some Complete Blood Count and red cell indices detected in palestinean families
 |
| |
|
Mean cell volume and MCHC were significantly decreased in individuals with HbA2 and HbF only not HbS which is insignificantly associated with MCV and MCHC.White blood cells were egually counted in individuals carrying HbA2, HbF, and HbS while platelets were significantly reduced in individuals carrying HbA2 and HbF only. Table 5 shows the significant increase of the range of detection of HbA2 by the electrophoresis rather than chromatographic technigue.Table 5. Comarison between the two methods used to determine of HbA2 (Gel Electrophoresis and Chromatography Ion Exchange Resin)
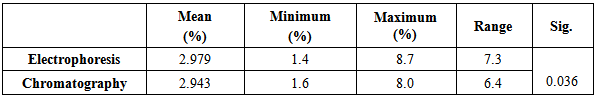 |
| |
|
Figure 1 shows the percentages of elevated HbA2 (> 3.7 %) among all families which is represented in separate bars. The percentage was highly increased in Torokman, Amarneh, Qalalweh and Zaqzooq familes. 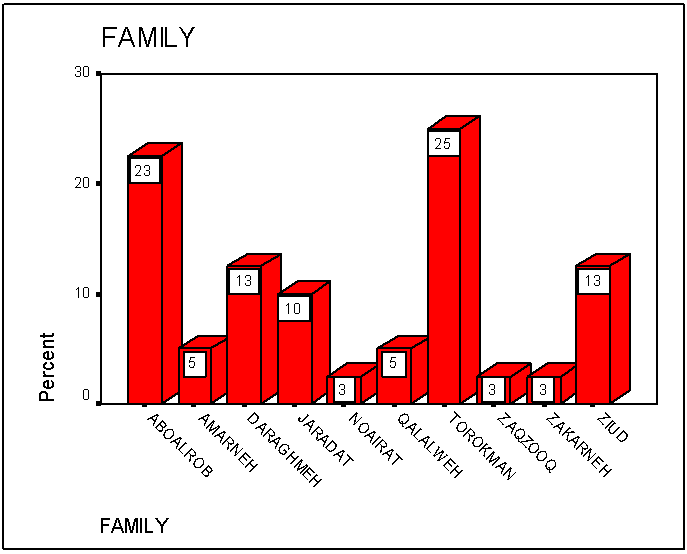 | Figure 1. Means of the concentration of elevated HbA2 (>3.7%) in the major families in Palestine |
Figure 2 shows the percentages of elevated HbF (>2.0%) in each family being represented in bars which was increased in Aboalrob and Torokman followed by Ziud, Amarneh, Daraghmeh, Qalalweh and Zakarneh.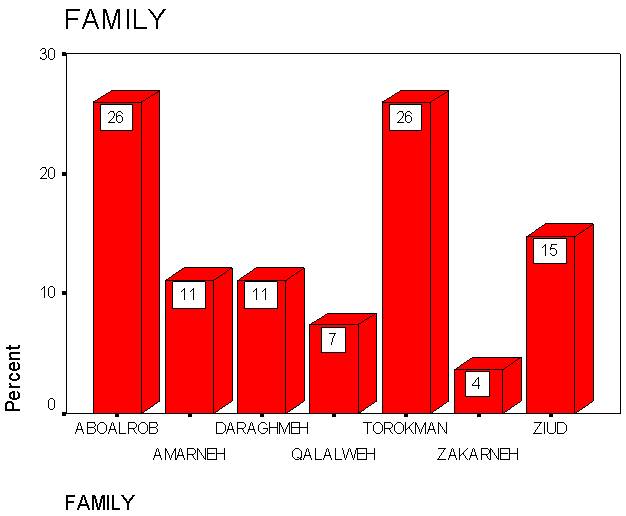 | Figure 2. Percentage of the elevated HbF concentration (>2.0%) in the major families in Palestine |
As shown in figure 3, HbS was detected in 6 families in Palestine; it was highly present in Qalalweh family followed by Noairat, Aboalrob, Zaqzooq and Amarneh families.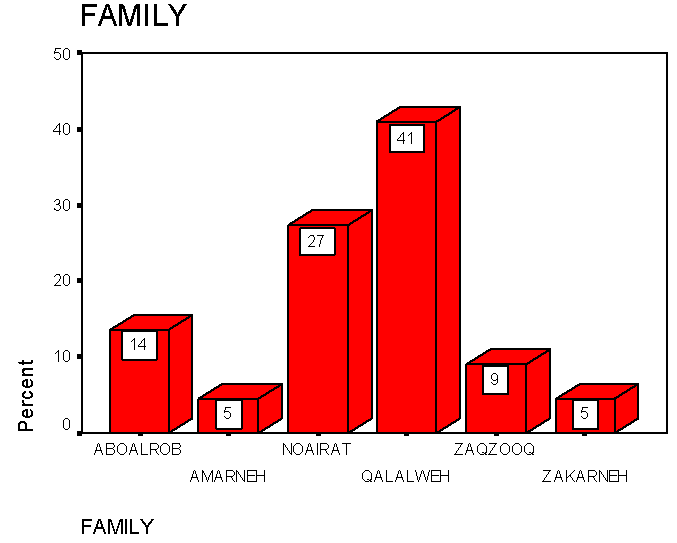 | Figure 3. The occurance of HbS in the major families in Palestine |
4. Discussion
Thalassaemias are a major health problem and poses real threat in Mediterrnian countries including Plaletine.The study was conducted in Jenin area in Palestine, where 200,000 people and more than 150 diagnosed Beta Thalassaemia major patients live. Therefore, it is essential to find out the prevalence of beta thalassaemia in order to reduce such risk. The study is important because informations and data about other abnormal Hb variants present in this area could be obtained. Moreover, the study recruits the major families present in this area, which represents most of populations in that region as it considered the mai source of the agricultural products in Palestine. The study showed that decreased level of HbA (<96%) was observed in 70 cases (27%) while elevated Hb A2 (>3.7%) in 40 cases (15.2%). Also reveals elevated HbF (>2.0%) in 27 cases (10%) and 22 cases of HbS (8%). The percentage of elevated HbA2 (>3.7%), varies among families and it was constituted the highest proportion in Torokman (25%), followed by Aboalrob (23%), Ziud and Daraghmeh (13%) each and of smaller percentages in the other families. On the other hand, the percentage of elevated HbF was highly detected in Torokman and Aboalrob families (26% each) followed by the other families. Elevated HbF in all families is close to the increase in HbA2 amomg them. This combination confirms the diagnosis of B-thalathemia among those individuals. There is obvious increase in HbA2 concentration among male individuals over females and such increase does not present in HbA, HbF and HbS. This is likely due to chance since no previously published data.It was obvious from the study that HbS is present in six families and it is highly increased in Qalalweh, Noairat and Aboalrob and less percentages in the other three families (Zakarneh, Amarneh and Zaqzooq), while does not present in the rest ‘Ziud, Daraghmeh, Jaradat and Torokman. The study shows no correlation between each of color of eye, color of hair, type of hair and length of individuals versus the mean concentration of HbA, HbA2, HbF and HbS. This can be explained by the fact that the genes responsible for these physical characters are located on different chromosomes; Haemoglobins variant genes are located on chromosomes 15 and 19 while eye, hair and tall genes located on choromosomes 11 and 16 [8]. No previous studies were found concerning this correlation.In this study, Hb, PCV, and RBCs are significantly reduced in individuals having HbA2 and HbF, because these Hb's result in hypochromic/microcytic profile contrary to HbAS which does not result in reduction of Hb, PCV and RBCs because it does not destruct RBCs except in special circumstances. This is agree to that reported by Hoffbrand AV et al [9, 10].The increase of the detection limit of HbA2 concentration measured by electrophoresis was significantly greater than that measured by Ion exchange chromatography method. This is agreeing with a previous study intended to compare between densitometry and chromatography and commented that densitometry is not sufficient and not satisfactory for diagnosing beta thalassaemia trait [11]. The study concluded that B Thalassaemia detected in jenin populations with variable percentages, being high in Torokman and followed by Abolarob, Ziud and Daraghmeh. Also, Sickle cell gene was present in three major families: Qalalweh, Noairat and Abolarob respectively. Therefore, the study recommended establishing a social protocol for marriage based on haematological investigations and official medical supervision to prevent Thalassemia major firstly. If the spouse is not a carrier, there is absolutely no chance that the children will have Thalassaemia Major, though they may have the Thalassaemia trait like their father. Second, if the spouse is a carrier, there is a 25% chance to that the child may have Thalassaemia Major. Moreover, the social protocol may includes mass education, mass screening, extended family screening of prenatal diagnosis and termination of pregnancy.
References
| [1] | Hunter R C, Hare A O, and Erim G, Diseases, Haematology: principles and procedures, 1993; 1sted.: 279-344. |
| [2] | Lewis SM, Bain B J and Bates I, investigation of abnormal haemoglobins and thalassaemia, Dacie and Lewis practical Haematology, 2001; 9th ed.: 231-268. |
| [3] | Steinberg M H, Forget BG, Higgs DR, Nagel RL, Disorders of Haemoglobin: Genetics, pathophysiology, and clinical management, Molecular mechanisms of beta thalassaemia, 2001: 252-276. |
| [4] | Stevens ML, Normal responses and Diseases of Erythrocytes. Stevens fundamentals of clinical haematology, 1997: 173-197. |
| [5] | Hoffbrand A Victor, Catovsky Daniel and Tuddenham Edward G.D. Haemoglobin and the inherited disorders of globin synthesis. Postgraduate Haematology. Black Well Publishing. Fifth edition 2005, Chapter6, pp 85. |
| [6] | Ciesla Betty. Microcytic Hypochromic Anemias, The Thalassemia Syndromes, Hematology in Practice; F A Davis 2007, pp74. |
| [7] | Moi P, Faa V, Marini MG, et al, Anovel silent beta thalassaemia mutation in the dstal CACCC box affects the binding and responsiveness to EKLF, Br J Haematol, 2004; 126: 881-884. |
| [8] | Ali Mohamed Siddig, Kadaru AG. In vitro processing of donor blood with Sulfadoxine-Pyrimethamine for eradication of transfusion-induced malaria. Am. J. Trop. Med. Hyg., 2005: 73(6), 1119–1123. |
| [9] | Dacie JV, Lewis SM. Practical hematology. 9th ed. Churchill Livingstone: Edinburgh; 2001:453. |
| [10] | Eiberg H, Mohr J. Assignment of genes coding for brown eye and brown hair color on chromosom 15. Euro. J Hum. Gen., 1996; 4:237 – 41. |
| [11] | Hoffbrand A V and Lewis S M, the Haemoglobinopathies, post graduate Haematology, 1981 2nd ed.: 190-228. |
| [12] | Hoffbrand A V, Pettit JE and Moss P A H, Genetic disorders of haemoglobin, Essential Haematology, 2001, 4th ed.: 71-90. |
| [13] | Schmidt RM, Ruchnogel DL and Necheles TF. Comparison of methodologies for Thalassaemia screening by HbA2 quantitation. J Lab Clin Med. 1975; 86(5): 873 – 82. |
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML