-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Medicine and Medical Sciences
p-ISSN: 2165-901X e-ISSN: 2165-9036
2013; 3(4): 57-60
doi:10.5923/j.ajmms.20130304.01
What Induces Diabetic Ketoacidosis or Lactic Acidosis in Diabetic Alcoholic Patients Complicated with Chronic Calcific Pancreatitis?
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLH. Yanai1, 2, H. Adachi1, H. Hamasaki1
1Department of Internal Medicine, National Center for Global Health and Medicine Kohnodai Hospital, Chiba, Japan
2Clinical Research Center, National Center for Global Health and Medicine Kohnodai Hospital, Chiba , Japan
Correspondence to: H. Yanai, Department of Internal Medicine, National Center for Global Health and Medicine Kohnodai Hospital, Chiba, Japan.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Diabetic ketoacidosis (DKA) and lactic acidosis (LA) are severe metabolic acidosis which develop in diabetic alcoholic patients. The insulin deficiency and elevation of glucagon leads to increased hepatic glucose output, and induces the release of free fatty acids (FFA) from adipose tissue, which are associated with the development of DKA. The insulin deficiency also plays a critical role in LA because pyruvate dehydrogenase complex (PDHc) requires insulin for activation, and increased FFA decreases PDH activity. Chronic calcific pancreatitis (CCP)-induced diabetes has been reported to be not prone to develop DKA because CCP leads to depletion of both insulin (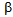 -cells) and glucagon-producing cells (
-cells) and glucagon-producing cells (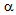 -cells) in pancreas. Further, to our knowledge, the development of LA in patients with CCP-induced diabetes has not ever been reported. We experienced alcoholic DKA and LA patients complicated with CCP-induced diabetes. The insulin deficiency is the critical factor for the development of both DKA and LA. FFA release from adipose tissue may be an important factor to determine the development of DKA. The absence of DKA in patient with LA may be due to extremely small volume of visceral adipose tissue. Although FFA is associated with the development of LA, a decreased activity of PDHc by insulin deficiency may be the most critical factor for the development of LA. Less insulin may be required to treat or prevent LA compared with DKA.
-cells) in pancreas. Further, to our knowledge, the development of LA in patients with CCP-induced diabetes has not ever been reported. We experienced alcoholic DKA and LA patients complicated with CCP-induced diabetes. The insulin deficiency is the critical factor for the development of both DKA and LA. FFA release from adipose tissue may be an important factor to determine the development of DKA. The absence of DKA in patient with LA may be due to extremely small volume of visceral adipose tissue. Although FFA is associated with the development of LA, a decreased activity of PDHc by insulin deficiency may be the most critical factor for the development of LA. Less insulin may be required to treat or prevent LA compared with DKA.
Keywords: Chronic Calcific Pancreatitis, Diabetic Ketoacidosis, Free Fatty Acids, Lactic Acidosis
Cite this paper: H. Yanai, H. Adachi, H. Hamasaki, What Induces Diabetic Ketoacidosis or Lactic Acidosis in Diabetic Alcoholic Patients Complicated with Chronic Calcific Pancreatitis?, American Journal of Medicine and Medical Sciences, Vol. 3 No. 4, 2013, pp. 57-60. doi: 10.5923/j.ajmms.20130304.01.
1. Introduction
- Diabetic ketoacidosis (DKA) and lactic acidosis (LA) are severe metabolic acidosis which develops in diabetic alcoholic patients[1]. The insulin deficiency and elevation of glucagon leads to increased release of glucose by the liver due to increased glycogenolysis and gluconeogenesis[2]. The insulin deficiency also leads to the release of free fatty acids (FFA) from adipose tissues, which are converted into ketone bodies, inducing DKA. The insulin deficiency also plays a critical role in LA because pyruvate dehydrogenase complex (PDHc) requires insulin for activation[3]. Increased FFA also decreases PDHc activity[4]. The PDHc is a mitochondrial matrix multienzyme complex that provides the link between glycolysis and the tricarboxylic acid cycle by catalyzing the conversion of pyruvate into acetyl-CoA[5]. Therefore, PDHc deficiency induces LA[5].Chronic calcific pancreatitis (CCP) is defined as a continuing inflammatory disease of pancreas characterized by irreversible patho-morphological changes including calcification, and is often associated with the loss of exocrine and/or endocrine function[6,7]. Although CCP leads to the development of diabetes[8], the CCP-induced diabetes has been reported to be not prone to develop DKA because CCP leads to depletion of both insulin (
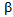 -cells) and glucagon-producing cells (
-cells) and glucagon-producing cells (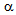 -cells) of the islets of Langerhans in pancreas[9]. Further, to our knowledge, the development of LA in patients with CCP-induced diabetes has not ever been reported. We experienced alcoholic DKA and LA patients complicated with CCP-induced diabetes. Here, we will discuss the clinical differences between patients with DKA and LA in the CCP-induced diabetes, and also discuss the possible pathological mechanisms to make this difference.
-cells) of the islets of Langerhans in pancreas[9]. Further, to our knowledge, the development of LA in patients with CCP-induced diabetes has not ever been reported. We experienced alcoholic DKA and LA patients complicated with CCP-induced diabetes. Here, we will discuss the clinical differences between patients with DKA and LA in the CCP-induced diabetes, and also discuss the possible pathological mechanisms to make this difference.2. Case Presentation
- Patient A and patient B presented with nausea and disturbed consciousness after drinking much alcohol drinks. Both patients were alcoholic. Blood gas analysis of patient A revealed severe metabolic acidosis (pH 6.99). Plasma glucose level (731 mg/dl) was extremely high and urinary ketones were strongly positive (Table 1). These data suggested that patient A developed DKA. Hydration and intensive insulin therapy promptly reduced urinary ketones, and ameliorated his consciousness and metabolic acidosis. Serum triglyceride and plasma glucose levels were also significantly decreased. Finally, his plasma glucose levels became to be around 150 mg/dl by the treatment with pre-mixed insulin 30/70 (5 units each before breakfast and dinner) and insulin glargine (10 units before sleep). Blood gas analysis of patient B showed acidosis (pH 7.337) and increased lactate levels (11.45 mmol/l; normal range, < 2.0 mmol/l). Patient B showed moderately high level of plasma glucose (265 mg/dl) and slightly positive urinary ketones, and also slightly elevated serum levels of total ketones (354
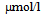 ) and 3-hydroxybutyrate (314
) and 3-hydroxybutyrate (314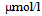 ) and normal level of acetoacetate (40
) and normal level of acetoacetate (40 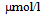 ), challenging the existence of severe ketoacidosis (DKA and alcoholic ketoacidosis) in patient B, and finally patient B was diagnosed as having alcoholic LA. Hydration and intravenous insulin infusion (5 units/day) promptly ameliorated his consciousness and metabolic acidosis. Serum lactate levels decreased to 3.16 and 1.21 mmol/l, at 5 and 45 hours after the treatment started, respectively. Plasma glucose levels at 5 and 45 hours after the treatment started was 303 and 213 mg/dl, respectively.Clinical and biochemical characteristics of CCP patients complicated with DKA and LA were shown in Table 1. Abdominal computed tomography (CT) of both patients showed the existence of CCP (Figure 1). Body mass index (BMI), plasma glucose, and serum levels of total cholesterol and triglyceride in patient B were significantly lower than those in patient A. Daily urinary C-peptide secretion and HbA1c in patient B was similar to those in patient A. Fasting serum glucagon level in patient B was significantly higher as compared with that in patient A. Daily dose of required insulin to treat for patient B was significantly smaller than that for patient A.Abdominal CT of patient B showed extremely small volume of visceral adipose tissue (26.7 cm2) (Figure 2).
), challenging the existence of severe ketoacidosis (DKA and alcoholic ketoacidosis) in patient B, and finally patient B was diagnosed as having alcoholic LA. Hydration and intravenous insulin infusion (5 units/day) promptly ameliorated his consciousness and metabolic acidosis. Serum lactate levels decreased to 3.16 and 1.21 mmol/l, at 5 and 45 hours after the treatment started, respectively. Plasma glucose levels at 5 and 45 hours after the treatment started was 303 and 213 mg/dl, respectively.Clinical and biochemical characteristics of CCP patients complicated with DKA and LA were shown in Table 1. Abdominal computed tomography (CT) of both patients showed the existence of CCP (Figure 1). Body mass index (BMI), plasma glucose, and serum levels of total cholesterol and triglyceride in patient B were significantly lower than those in patient A. Daily urinary C-peptide secretion and HbA1c in patient B was similar to those in patient A. Fasting serum glucagon level in patient B was significantly higher as compared with that in patient A. Daily dose of required insulin to treat for patient B was significantly smaller than that for patient A.Abdominal CT of patient B showed extremely small volume of visceral adipose tissue (26.7 cm2) (Figure 2).
|
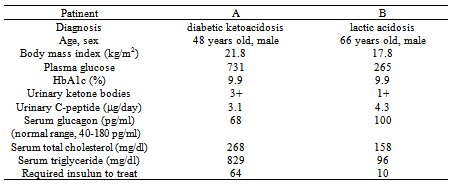
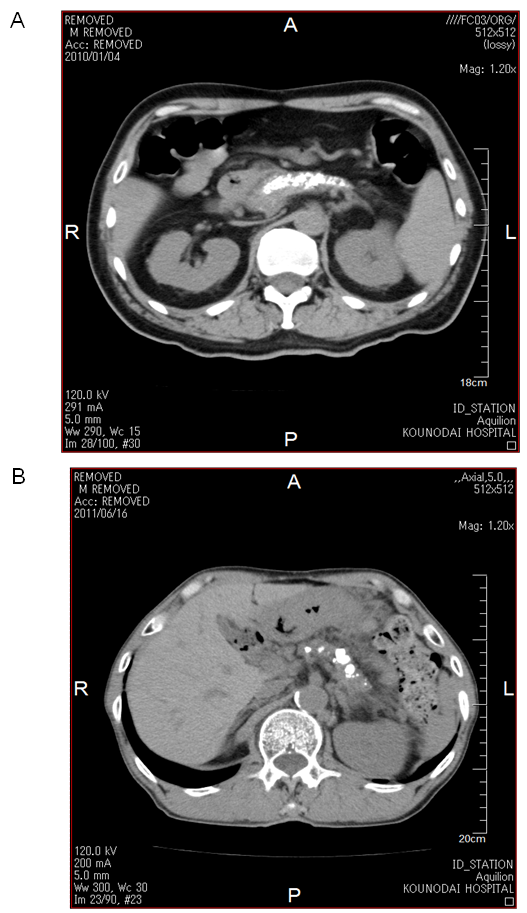 | Figure 1. Computed tomography of abdomen of patient A (A) and patient B (B) |
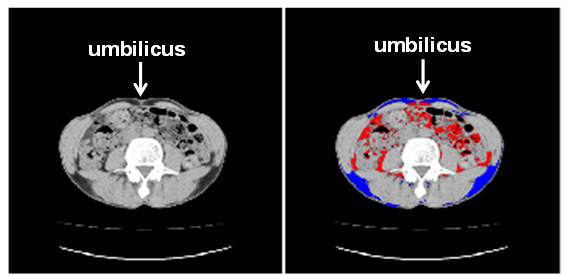 | Figure 2. Subcutaneous (blue area) and visceral (red area) fat in patient B detected by abdominal computed tomography |
3. Discussion
- CCP is a destructive inflammatory disease of the pancreas[6]. Although the etiology of CCP is multifactorial, alcoholism plays a significant role in adults[10]. Our patients were also alcoholic. The recurrence of CCP leads to decline of endocrine and exocrine pancreatic function[6]. Therefore, diabetes mellitus is frequently complicated with CCP[6]. Surgical therapy has been reported to be effective to improve symptoms and quality of life for patients with pancreatic duct stone (PDS) and CCP, and also has been reported to improve pancreatic function[6,11,12]. However, the influence of surgical therapy on the onset of diabetes and glucose metabolism has been remained unknown. Very recently, Liu BN, et al studied the surgical procedures for PDS in patients with CCP, and found that the surgical therapy reduced abdominal pain but induced diabetes in several patients[13], indicating the necessity for further accumulation of studies about diabetic patients complicated with CCP.DKA is characterized by insulin deficiency, which induces the release of great amounts of FFA from adipose tissue, and by glucagon excess, which regulates the conversion of FFA into ketone bodies in the liver[2]. Hormone-sensitive lipase (HSL) is the enzyme responsible for the hydrolysis of triacylglycerol from the lipid droplet of adipocytes into glycerol and FFA[14]. Insulin deficiency increases expression of HSL via pre-translational mechanisms[15]. Glucagon also enhances lipolysis and stimulates mitochondrial conversion of FFA into ketone bodies[16]. Insulin normally blocks ketogenesis by inhibiting the carnitine palmitoyltransferase (CPT)-mediated transport of FFA derivatives into the mitochondria, but insulin deficiency decreases concentrations of malonyl CoA which inhibits FA oxidation by inhibiting CPT[17]. Glucagon also activates CPT by phosphorylation through cAMP-dependent protein kinase[18]. Therefore, the existence of insulin deficiency, elevation of glucagon and increased release of FFA from adipose tissue are crucial for the development of DKA. As CCP leads to depletion of both insulin (
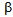 -cells) and glucagon-producing cells (
-cells) and glucagon-producing cells (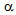 -cells) of the islets of Langerhans in pancreas, diabetes due to CCP are usually not to prone to develop DKA, because excess glucagon do not develop under the insulin deficient condition[9]. However, our patient A preserved glucagon secretion capacity in spite of severely reduced insulin secretion capacity, which may induce the onset of DKA. Why did not DKA develop in the patient B? Insulin deficiency in patient B is as severe as that in patient A, and serum glucagon level in patient B is higher than that in patient A. Patient B seems to be more likely to develop DKA compared with patient A. BMI, serum total cholesterol and triglyceride levels were significantly lower in patient B compared with patient A. Abdominal CT showed extremely small volume of visceral adipose tissue. FFA release from adipose tissue may be an important factor to determine the development of DKA. The absence of DKA in patient B may be due to extremely small volume of visceral adipose tissue. Elevations in plasma glucagon concentration have been reported to be augment hepatic ketogenesis in patients with type 1 diabetes when simultaneous elevations in serum FFA are present[19], suggesting the importance of FFA for glucagon-mediated development of DKA. Increased FFA decreases PDHc activity[3]. Although FFA is associated with the development of LA, FFA metabolism may be significantly reduced in patient B, which is supported by normal serum triglyceride levels and very small adipose tissue. A significantly decreased activity of PDHc by insulin deficiency may be the most critical factor for the development of LA in patient B. Less insulin may be required to treat or prevent LA compared with DKA. We have to mention the limitation of our study. The main limitation is the small number of patients. Therefore, our concept and/or hypothesis are not lead by pathophysiological proof, but, by mainly speculation. In conclusion, the insulin deficiency is the critical factor for the development of both DKA and LA. FFA release from adipose tissue may be an important factor to determine the development of DKA. The absence of DKA in patient with LA may be due to extremely small volume of visceral adipose tissue. Although FFA is associated with the development of LA, a reduced activity of PDHc by insulin deficiency may be the most critical factor for the development of LA. Less insulin may be required to treat or prevent LA compared with DKA. The association between the onset of DKA or LA and the balance of preserved functions of
-cells) of the islets of Langerhans in pancreas, diabetes due to CCP are usually not to prone to develop DKA, because excess glucagon do not develop under the insulin deficient condition[9]. However, our patient A preserved glucagon secretion capacity in spite of severely reduced insulin secretion capacity, which may induce the onset of DKA. Why did not DKA develop in the patient B? Insulin deficiency in patient B is as severe as that in patient A, and serum glucagon level in patient B is higher than that in patient A. Patient B seems to be more likely to develop DKA compared with patient A. BMI, serum total cholesterol and triglyceride levels were significantly lower in patient B compared with patient A. Abdominal CT showed extremely small volume of visceral adipose tissue. FFA release from adipose tissue may be an important factor to determine the development of DKA. The absence of DKA in patient B may be due to extremely small volume of visceral adipose tissue. Elevations in plasma glucagon concentration have been reported to be augment hepatic ketogenesis in patients with type 1 diabetes when simultaneous elevations in serum FFA are present[19], suggesting the importance of FFA for glucagon-mediated development of DKA. Increased FFA decreases PDHc activity[3]. Although FFA is associated with the development of LA, FFA metabolism may be significantly reduced in patient B, which is supported by normal serum triglyceride levels and very small adipose tissue. A significantly decreased activity of PDHc by insulin deficiency may be the most critical factor for the development of LA in patient B. Less insulin may be required to treat or prevent LA compared with DKA. We have to mention the limitation of our study. The main limitation is the small number of patients. Therefore, our concept and/or hypothesis are not lead by pathophysiological proof, but, by mainly speculation. In conclusion, the insulin deficiency is the critical factor for the development of both DKA and LA. FFA release from adipose tissue may be an important factor to determine the development of DKA. The absence of DKA in patient with LA may be due to extremely small volume of visceral adipose tissue. Although FFA is associated with the development of LA, a reduced activity of PDHc by insulin deficiency may be the most critical factor for the development of LA. Less insulin may be required to treat or prevent LA compared with DKA. The association between the onset of DKA or LA and the balance of preserved functions of 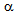 -cells and
-cells and 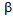 -cells in CCP remains unknown. The precise mechanisms for FFA-mediated development of DKA and LA also remain to be elucidated. Further studies, preferably with larger numbers of subjects, will be needed in the future.
-cells in CCP remains unknown. The precise mechanisms for FFA-mediated development of DKA and LA also remain to be elucidated. Further studies, preferably with larger numbers of subjects, will be needed in the future.