-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Dermatology and Venereology
p-ISSN: 2332-8479 e-ISSN: 2332-8487
2020; 9(2): 21-26
doi:10.5923/j.ajdv.20200902.02

Clinical and Histopathological Evaluation of Pigmented Morphea with New Insight in Relation to Etiopathogenesis of the Disease
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLKhalifa E. Sharquie1, Adil A. Noaimi1, Eman T. Abdulqader2, Wesal K. Aljanabi2
1Department of Dermatology, College of Medicine, University of Baghdad, Iraqi and Arab Board for Dermatology and Venereology, Baghdad Teaching Hospital, Medical City, Baghdad, Iraq
2Dermatology Center, Medical City, Baghdad, Iraq
Correspondence to: Wesal K. Aljanabi, Dermatology Center, Medical City, Baghdad, Iraq.
| Email: |  |
Copyright © 2020 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Background Morphea is an autoimmune disease where there are early erythematous patches with or without lilac border and ending with sclerosis. While in Iraqi population, morphea patients present with pigmented patches that might stay for a long time before changing into sclerotic ivory white plaques. The objective of this study is to do clinical and histopathological evaluation of different stages of morphea. This is a descriptive study that was conducted in Medical city, Baghdad, where 100 patients with morphea were included. Clinical and histopathological evaluation was carried out. Results Their ages ranged from 2 – 60 years old with M±SD=26.24±15.345, and female to male ratio 1.5: 1. In all cases, the lesion started as pigmented, non-indurated areas that might stay for several months and even years before progressing into ivory white sclerotic plaques. The histopathological changes in morphea could be classified into early pigmented phase where there was mild acanthosis, increase in basal melanosis, vascular dilatation and massive deposition of collagen together in the dermis and panniculus. While in late cases especially with sclerosis there was in addition thinning and atrophy of epidermis with basal liquefaction plus homogenization and hyalinization of collagen in papillary dermis simulating lichen sclerosus. Conclusion this study shows striking clinical and histopathological findings as all cases presented with pigmented patches that are compatible with histopathological changes where there was collagen deposition with basal hyper melanosis in early cases, with thinning of epidermis and homogenization of papillary dermis in late cases.
Keywords: Pigmented morphea, Scleroderma, Histopathology
Cite this paper: Khalifa E. Sharquie, Adil A. Noaimi, Eman T. Abdulqader, Wesal K. Aljanabi, Clinical and Histopathological Evaluation of Pigmented Morphea with New Insight in Relation to Etiopathogenesis of the Disease, American Journal of Dermatology and Venereology, Vol. 9 No. 2, 2020, pp. 21-26. doi: 10.5923/j.ajdv.20200902.02.
Article Outline
1. Introduction
- Morphea was a rare skin problem in 1970s in Iraqi population. Since 1980s the disease was increasing and now it is not uncommon disease. The etiology of the disease is not well elucidated but the clinical and laboratory findings are in favor of autoimmune reaction such as elevation of antinuclear antibody, association with diseases such as lupus erythematosus, rheumatoid arthritis (RA), systemic sclerosus and transverse myelitis [1]. Resolution and improvement with immunosuppressive therapy also support this theory [2]. It is currently thought that patients who develop morphea have an underlying genetic predisposition to this condition, and then are exposed to an environmental factor that initiates the inflammatory and fibrotic cascades [3]. Some theories proposed that injury to the vascular endothelium during the inflammatory stage stimulates the release of cytokines that upregulate the expression of vascular adhesion molecules, including vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and E-selectin. These adhesion molecules facilitate the recruitment of especially T-lymphocytes that are capable of producing profibrotic cytokines mainly IL-4, IL-6, and TGF-beta, and may contribute to the development to sclerosis. Genetic and multiple environmental factors like infections or environmental exposures also have been proposed as contributors to disease expression [4].The ordinary clinical picture of morphea usually present as asymptomatic slightly elevated, erythematous or violaceous, somewhat edematous plaque that undergoes centrifugal expansion then becomes sclerotic, scar-like tissue. Depending on the depth of the sclerosis, the skin becomes progressively indurated. Centrally, it can acquire a shiny white color, and peripherally, a violaceous or lilac ring. Post inflammatory hyperpigmentation often dominates over the white sclerosis as the lesions mature [5]. There are many varieties of morphea: Plaque type as a single or multiple oval to round lesions limited that may be superficial or deep, linear morphea that may involve the head (en coup de sabre) or the extremities or even the trunk, generalized morphea as coalescent plaque ≥4 plaques in at least 2 of 7 anatomic sites. (head–neck, right/left upper extremity, right/left lower extremity, anterior/posterior trunk) including pan sclerotic morphea that involve the majority of body surface area except the tips of the fingers and toes affecting the skin and may extend to the bone, and mixed type of morphea that is composed of combination of previous subtypes [6]. Other rare types are: guttate morphea [7], progressive facial hemi atrophy [8], bullous morphea [9], atrophoderma of pasini and pierini [10], keloidal or nodular morphea [11] and morphea profundus or deep morphea [12]. The extra cutaneous involvement in morphea is considered to be extremely unusual by many authors [13].The histopathology of the disease is not well elucidated in medical literature as the only mentioned changes are thickened collagen bundles in the dermis and inflammatory cell infiltrate between the collagen bundles and around blood vessels, which may extend into the subcutaneous fat and eccrine glands in early lesions, while the late lesions show a paucity of inflammatory cells, extension of the collagen bundles in the subcutaneous tissue and replacement of fat cells and atrophy of the glands [14].The objective of the present work is to do extensive clinical and histopathological evaluation of different stages of morphea including the pigmented and sclerotic phase.
2. Patients and Methods
- The current study is a descriptive, clinical and histopathological work that was carried out in the Center of Dermatology, Medical City, Baghdad-Iraq from October 2015 to October 2017. A total of 100 patients were included in this study, all of them had morphea in different sites of the body.Full history was taken from all the patients including the following: age, sex, occupation, complaint of the patients like itching and burning, duration of disease, type and color of lesion, associated systemic symptoms, associated autoimmune disease, medical history, surgical history and drug history.Clinical examination was done regarding the type, colour and the site of the lesions. Biopsy of the skin was taken from 20 patients of different clinical stages. Hematoxylin and eosin stain in addition to Fontana Masson stain was done for all these biopsies. Ipad air 2 (8 megapixel) and Iphone 7 (12 megapixel) plus cameras were used for photography.
3. Results
- A total number of 100 patients with morphea were seen, their ages ranged between 2 – 60 years mean ± SD = 26.24 ± 15.345. There were 60 females (60%) and 40 males (40%) with female to male ratio 1.5: 1. Patients were divided into the following age groups:• Ages ≤ 18 years were 39 patients (39%).• Ages between 18-, 45 years were 43 patients (43%).• Ages ≥ 45 years were 18 patients (18%).Accordingly, it was a disease of children and young people.
3.1. The Following Types of Morphea were Seen
- • Circumscribed morphea, fig. 1, (including the ordinary localized plaque type): 61 patients (61%) had this type, 34/61 females (55.73%) and 27/61 males (44.26%).
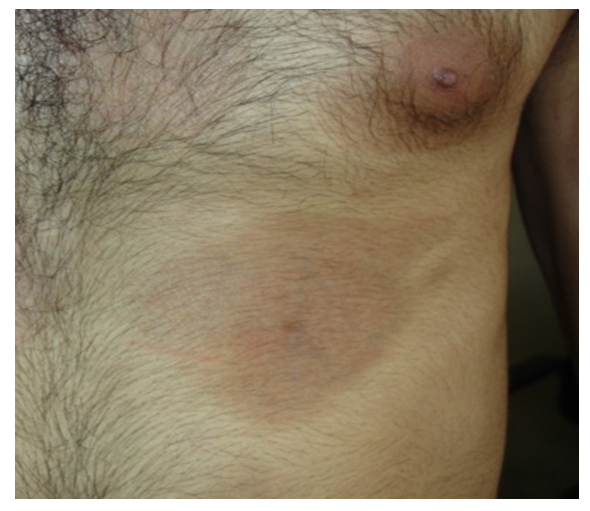 | Figure 1. Thirty years old male with early-pigmented circumscribed morphea on the trunk |
|
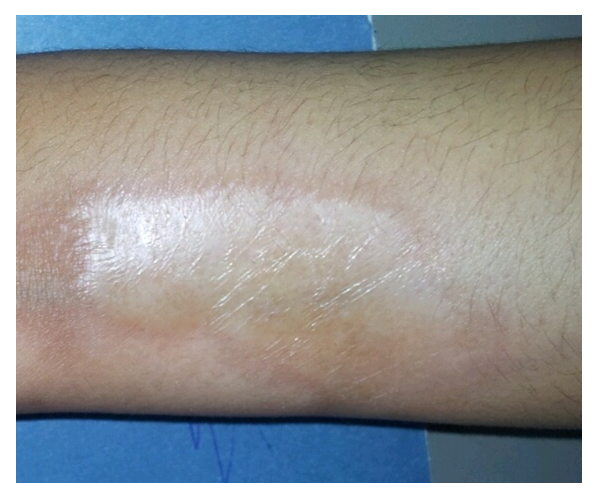 | Figure 2. Fifteen years old male with ivory sclerotic circumscribed lesion with lilac border on the left forearm |
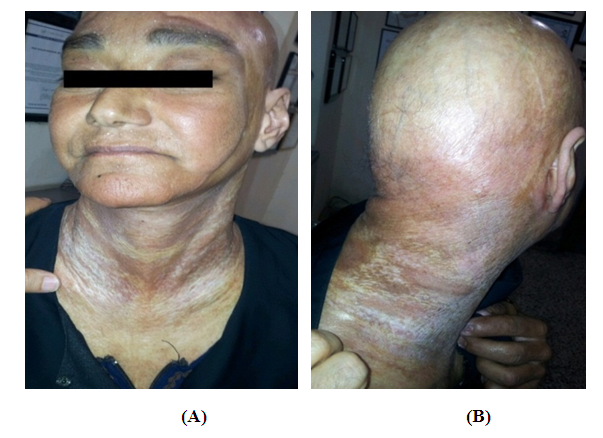 | Figure 3. (A and B) Forty years old, female showing severe generalized sclerotic morphea all over the body but sparing the face, hands and feet |
3.2. According to the Colour and Induration of the Lesion at Time of Presentation, Patients were Divided into Following Groups
- • Patients had pigmented non indurated lesions only: 44 patients (44%).• Patients had indurated lesions with ivory color only: 14 patients (14%).• Patients had mixed color of the two previous types: 28 patients (28%).• Patients had almost skin color non indurated lesions with atrophy: 14 patients (14%).In all cases, lesions started as macular-pigmented non itchy areas that might stay for several months to several years before they progress to ivory white sclerotic areas still some areas remain pigmented for a long time without changing into ivory white color, fig. 4. Twenty patients (20%) had lilac border mostly seen in the sclerotic plaques.
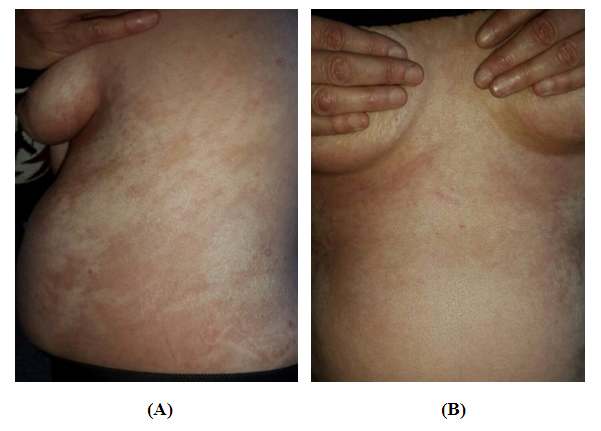 | Figure 4. (A and B) Forty-five years old female showing generalized pigmented morphea changing into whitish sclerotic area involving the trunk |
3.3. The Histopathological Changes were as Follow
- • In pigmented patches of early cases of morphea, fig. 5 & 6, there was mild acanthosis, normal dermal-epidermal junction, and obvious melanosis of epidermis that was mainly located in the basal layer, sometimes extended to the whole epidermis with normal papillary dermis but there was a slight to moderate perivascular lymphocytic infiltrate both superficial and deep without any melanophages. In addition, there were massive collagen depositions in the reticular dermis that might extend downward to occupy panniculus.
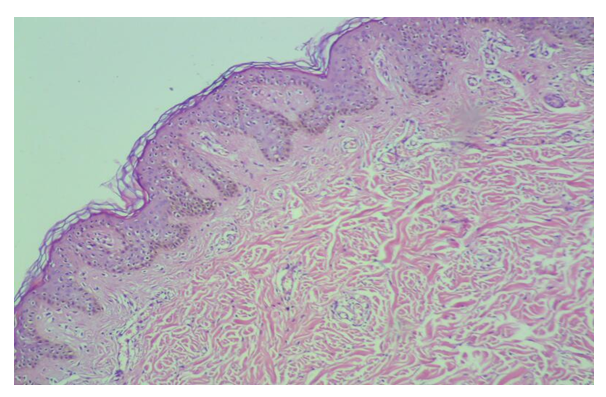 | Figure 5. Early pigmented lesion with epidermal acanthosis, basal melanosis, vascular dilatation, perivascular infiltrate and increased collagen deposition |
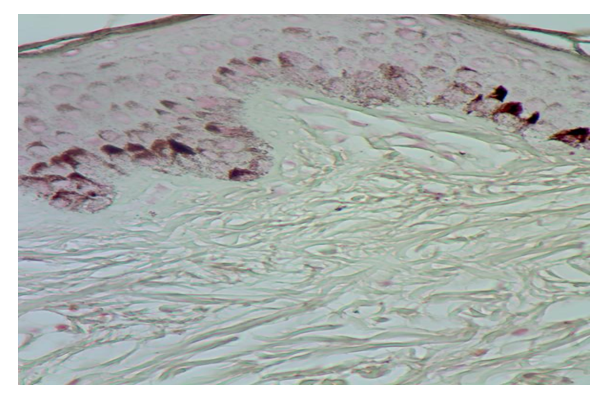 | Figure 6. Early pigmented lesion with Fontana Masson stain showing epidermal acanthosis, prominent basal and suprabasal melanosis extend up to the granular layer with excessive collagen deposition |
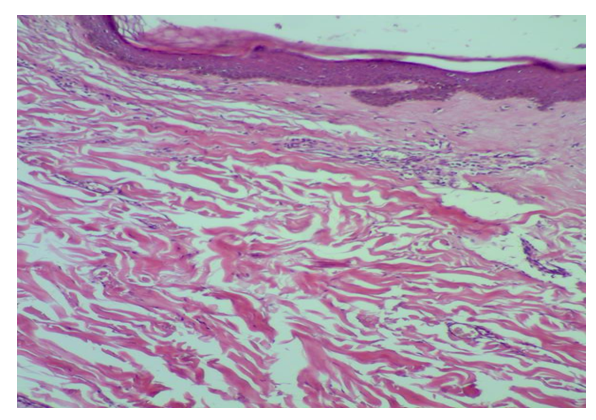 | Figure 7. Late pigmented lesion with normal epidermis, basal melanosis, hyalinization of papillary collagen, perivascular infiltrate and increased collagen deposition |
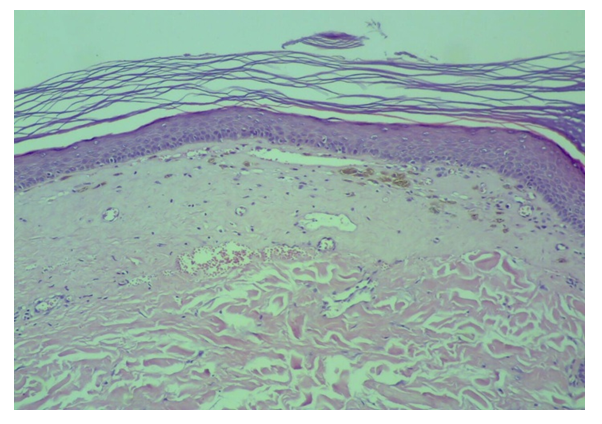 | Figure 8. Late sclerotic lesion with epidermal atrophy, hyalinization of papillary collagen, vascular dilatation, sparse perivascular infiltrate, melanophages and increased collagen deposition, simulating the picture of lichen sclerosus et atrophicus |
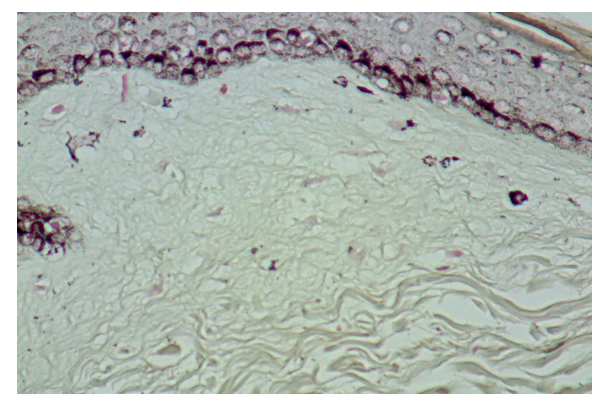 | Figure 9. Late sclerotic lesion with Fontana Masson stain showing atrophy of the epidermis with basal melanosis and few melanophages in the hyalinized papillary dermis |
4. Discussion
- In Iraq, the incidence of morphea is increasing overtime since 1980. While in a survey from Olmsted County, Minnesota, from 1960 to 1993, during this period, the annual incidence rate was 27 per million inhabitants [15].According to our study female to male ratio was 1.5:1 and all ages can be affected but mostly the young age group. In other studies, it was noticed that morphea occurs 2.6–6 times more frequently in women than in men [16]. All ages are affected, while the peak incidence occurring between 20 and 40 years of age [17].The clinical picture of Iraqi patients is different to what has been published in medical literature as in all varieties of morphea the patients presented with pigmented non-indurated patches that might stay for several months or even years before changing into ivory sclerotic areas with or without lilac border.In other literature, morphea usually begins as a red or purple area of skin that then becomes thickened and white. The thick white areas usually thin out over time and turn brown [3].Regarding the pathology of morphea in the present work there were two phases mainly showed prominent histopathological changes as follow: in the early-pigmented phase, there was mild acanthosis, melanosis of the epidermis especially the basal layer, normal dermo-epidermal junction, and normal papillary dermis with slight to moderate superficial and deep perivascular lymphocytic infiltrate but without melanophages. In the reticular dermis there was massive collagen deposition that might extend into the panniculus. While in the late sclerotic lesion in the second phase there was atrophy of the epidermis with decrease in basal melanosis and focal damage to the basal layer sometimes making a cleft in addition to hyalinization of collagen in the papillary dermis with few melanophages and sparse perivascular infiltrate. In all biopsies, vasodilatation in superficial and deep vessels was obvious this considered as new finding was not mentioned in other literature that documented morphea might present with histological findings very similar to systemic sclerosis [18].In the early phase, the reticular dermis typically shows dense perivascular and peri adnexal inflammatory infiltrates, sometimes depending on the case extending into the subcutaneous tissue [19].In late stage, it is characterized by intense fibrosis in the dermis, which progressively substitutes the adipose panicle. The inflammatory infiltrate in this stage is absent or discreetly confined around adnexae that already show atrophy. With the evolution of the disease, the tendency is that adnexae will be replaced by fibrosis [20].There are two theories that explain the pigmentation in cases of morphea, the first one there is injury to blood vessels similar to the changes in melasma after dermal inflammation hence there is significant increase in both the number and size of dermal blood vessels after dermal inflammation, and upregulated expression of vascular endothelial growth factor (VEGF), in the lesional skin compared to the perilesional normal skin. It has been speculated that UV irradiation induces an angiogenic switch, associated with the upregulation of proangiogenic factors such as VEGF, basic fibroblast growth factor (b-FGF), and interleukin-8. VEGF is the major putative angiogenic factor and it seems to enhance melanogenesis by interaction with VEGF receptors present in epidermal keratinocytes followed by release of mediators, most importantly metabolites of arachidonic acid, and plasminogen from the proliferated vessels [21].The other possibility as there is stimulation of fibroblast to produce collagen fibers In addition these cells secrete cytokines, the most important one is IL8 (angiogenic factor) that leads to angiogenesis. [22] Recently, fibroblasts have also been shown to secrete growth-related oncogene(s), interferon-γ inducible protein (IP-10), platelet factor-4 (PF-4), epithelial neutrophil activating protein-78, monocyte chemoattractant proteins (MCP-1,-2,and-3), macrophage inflammatory proteins (MIP), regulated on activation normal T cell expressed and secreted (RANTES), Prostaglandin E2, vascular endothelial growth factor (VEGF), stem cell factor (SCF) and alpha melanocyte stimulating hormone (αMSH), all are released after proinflammatory stimuli. Many of these cytokines serve for recruitment of immune cells and some of them act for angiogenesis [22].In addition, alpha melanocyte stimulating hormone produced by endothelial cells and fibroblasts that acts on melanocortin receptors on different cells like keratinocytes, melanocytes even on endothelial cells and fibroblasts [23]. These cytokines might act directly on basal melanocyte to cause release of excessive amount of melanin to be deposited in keratinocytes of the epidermis that is why the morphea lesions are mostly pigmented.
5. Conclusions
- Patients with morphea often presented with pigmented non indurated patches that might stay for several months to several years without progressing to white ivory sclerotic plaques. The histopathology of disease was severe collagen deposition with mild acanthosis of the epidermis and associated with basal melanosis. While in the late sclerotic phase there was in addition, atrophy of epidermis with basal liquefaction and homogenization of papillary dermis thus simulating lichen sclerosus et atrophicus. In all cases, there was dilatation of superficial and deep plexus of the dermis. The etiopathogenesis of the disease according to the present work could be summarized as injury to fibroblasts and to the endothelium of blood vessels causing release of different types of cytokines, hence followed by pigmentation of the epidermis and collagen deposition in the dermis.
Source of Funding
- Covered by Iraqi board for medical specialization.