-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Dermatology and Venereology
p-ISSN: 2332-8479 e-ISSN: 2332-8487
2019; 8(3): 41-44
doi:10.5923/j.ajdv.20190803.02

Erythrokeratoderma Variabilis et Progressiva in Iraqi Population
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLKhalifa E. Sharquie1, Fatema A. Al-Jaralla2
1Department of Dermatology, College of Medicine, University of Baghdad, Iraqi and Arab Board for Dermatology & Venereology, Baghdad Teaching Hospital, Medical City, Baghdad, Iraq
2Departments of Dermatology, College of Medicine, Baghdad University, Baghdad, Iraq
Correspondence to: Khalifa E. Sharquie, Department of Dermatology, College of Medicine, University of Baghdad, Iraqi and Arab Board for Dermatology & Venereology, Baghdad Teaching Hospital, Medical City, Baghdad, Iraq.
| Email: |  |
Copyright © 2019 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Erythrokeratoderma variabilis et progressiva (EKVP) are heterogeneous inherited group of rare keratinising skin diseases characterized by well demarcated fixed hyperkeratotic plaques and transient erythematous patches. EKVP are a disorders caused by the mutation in the genes encoding connexin-31 (GJB3) and connexin-30.3 (GJB4), the components of gap junction, in an autosomal dominant and rarely in recessive manner. More than 200 cases are reported in patients with diverse genetic backgrounds. EKVP has been reported worldwide and most cases were whites of Northern and Middle European origin, but it also occurs in African Americans and Asians. Both sexes are affected equally. Herein, we report a total of seven cases of Iraqi patients with EKVP that appeared within the first year of life, aiming to shed light on this rare skin problem in order to help dermatologists to establish the right diagnosis and to keep EKVP included in differential diagnosis of other skin disorders that might have some similar clinical features.
Keywords: Erythrokeratoderma Variabilis et Progressiva, Iraq
Cite this paper: Khalifa E. Sharquie, Fatema A. Al-Jaralla, Erythrokeratoderma Variabilis et Progressiva in Iraqi Population, American Journal of Dermatology and Venereology, Vol. 8 No. 3, 2019, pp. 41-44. doi: 10.5923/j.ajdv.20190803.02.
1. Introduction
- The clinically and genetically heterogeneous group of erythrokeratodermas encompasses several rare genetic skin disorders, including autosomal dominant erythrokeratodermia variabilis (EKV) and progressive symmetric erythrokeratoderma (PSEK), as well as autosomal dominant spinocerebellar ataxia with erythrokeratodermia. The skin lesions of EKV and PSEK show many similarities, and a small subset of patients with features previously considered to be PSEK share the underlying cause of EKV. Therefore, it was recently proposed to classify this disorder as erythrokeratoderma variabilis et progressiva (EKVP) [1]. EKVP presents with migratory erythema and fixed hyperkeratotic plaques [2]. Lesions commonly occur in the early stage of life. The lesions are hyperkeratotic and well-marginated and have a tendency to become confluent. Lesions are usually distributed on the extensor surface of extremities, buttocks, and face while the erythematous lesions are transitory and show variation with stress and heat [3].Herein, we report a total of seven cases of Iraqi patients with EKVP.
2. Clinical Study
- A total of seven patients, four females and three males were studied between the years 2013 and 2019. The mean age of patients was 21.75 years (range 1.5-42). All the studied cases had brownish and red rashes that had started since childhood. The red lesions appeared from time to time, initially spread centrifugally, worsened with physical activities and heat, and disappeared spontaneously, however, the brownish lesions on the trunk and extremities (and face in one patient) did not disappear. The patients were seen by different physicians and were treated with various antifungal treatments that were not beneficial. The positive family history could not be delineated well. No systemic diseases were detected in all cases.The dermatological examination showed brown erythematous, annular and geographic plaques located on the anterior part of chest, arms, legs, abdomen and axillary region in two patients in so called erythrokeratoderma variabilis. These two patients had in addition fixed hyperkeratotic plaques Figure 1 and 2. While the other five patients had features of progressive symmetric erythrokeratoderma as these patients had fixed slowly progressive hyperkeratotic plaques on different parts of the body including the face in one child Figure 3 and 4. No lesions were detected on the palms, soles, nails, hair, or mucosa. The histopathological examination of the hyperkeratotic plaque showed basket like hyperkeratosis with papillomatosis and acanthosis while the dermis showed mild superficial perivascular lymphocytic infiltration Figure 5.No fungal elements were detected by potassium hydroxide preparations in any patient. The clinical and histopathological findings were consistent with EKVP.One patient (fig 3) was treated with oral isotretinoin and the rash was completely gone within the first three weeks but the patient should be kept on maintenance oral reduced isotretinoin dose to keep the remission state.
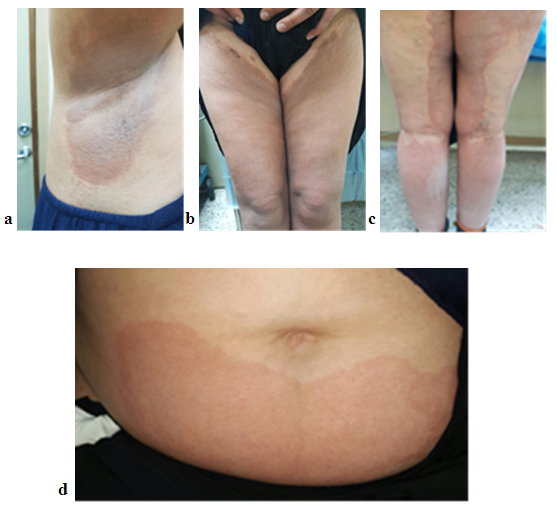 | Figure 1. (a, b, c, and d) 42 years woman with PSEK showing multiple well defined erythematous patches of right axilla, lower extremities and abdomen |
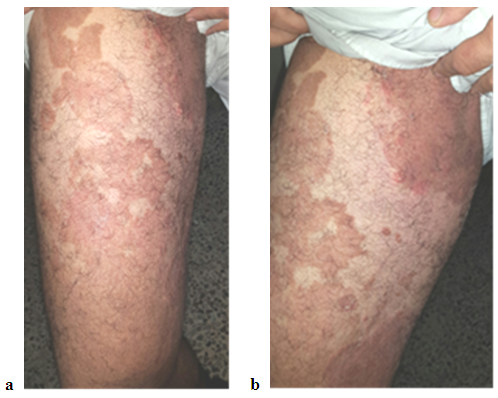 | Figure 2. (a and b) showing involvement of the right thigh and groin with well-defined red- brownish plaques in a 38 year old man with PSEK |
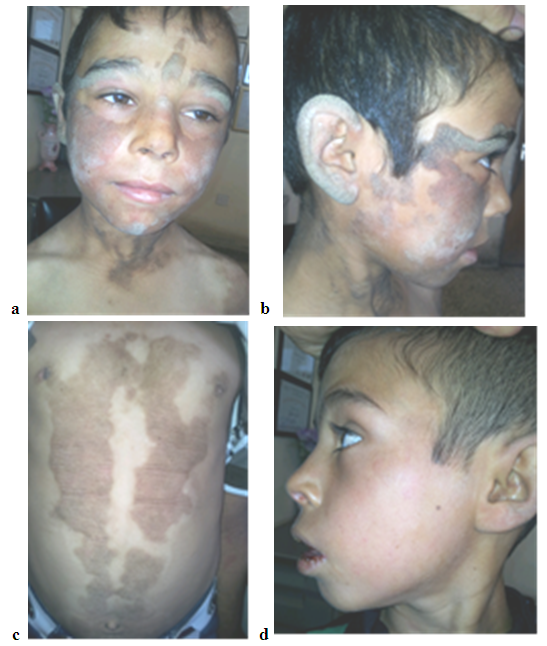 | Figure 3. (a, b and c) showing involvement of the face and trunk with brownish hyperkeratotic plaques in a 9 year old boy with PSEK Clearing of the lesions after 3 weeks of oral iostretinoin |
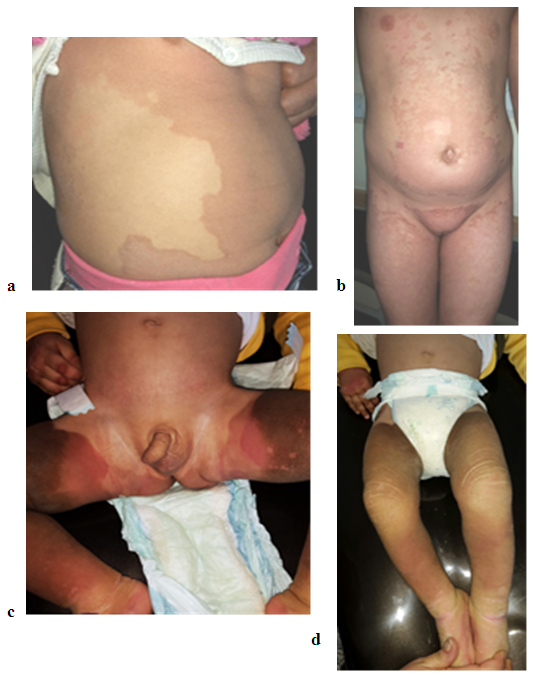 | Figure 4. (a) showing multiple well defined erythematous- brownish hyperkeratotic patches in a four years old girl with PSEK, (b) involvement of the trunk, thighs, genital area with erythematous patches in 7 years girl with EKV, (c and d) 18 months boy with EKV |
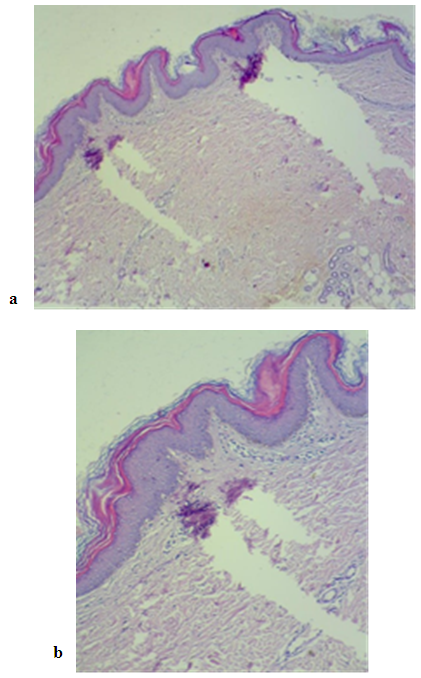 | Figure 5. (a and b) Microscopic view showing basket weave hyperkeratosis with underlying compact hyperkeratosis, acanthosis with slight papilomatosis. Mild superficial perivascular lymphocytic infiltrate was also noticed H & E stain × 40 (a) H & E stain ×100 (b) |
3. Discussion
- EKVP is characterized by the coexistence of two distinct morphologic features: fixed progressive hyperkeratosis and transient erythema, albeit one of these features may predominate. de Buy Wenninger recognized and described the first cases of erythrokeratodermia variabilis in the Netherlands in 1907 [4]. In 1925, Mendes da Costa presented a detailed clinical description of the disease in a mother and daughter, reviewed eight similar cases that were previously published, and coined the name "erythro- et keratodermia variabilis." [5]. During the next decades, multiple case reports emerged in the Northern European literature, including a study of 33 affected members of a Dutch family and 29 affected persons in 5 generations of a Swiss family [6]. In 1964, Barsky and Bernstein reported the first case in the American literature [7]. The first family in which affected members had features of either EKV or PSEK was reported in 1991 [8].Erythrokeratodermia variabilis et progressiva (EKVP) is rare, and its accurate prevalence is not known. More than 200 cases are reported in patients with diverse genetic backgrounds. EKVP has been reported worldwide. Most cases were whites of Northern and Middle European origin, but erythrokeratodermia variabilis also occurs in African Americans and Asians. Up to our knowledge, we are the first to report EKVP cases in Iraq.EKV shares many clinical features with PSEK. Since transient erythematous patches are subtle or absent in some EKV patients, especially during adulthood, it has been suggested that EKV and PSEK represent different clinical presentations of the same disorder. The term Erythrokeratoderma variabilis et progressive was proposed to encompass both entities [9].No specific therapy is available for EKVP. The use of topical agents in the management depends on the symptoms and focuses on hydration, lubrication, and keratolysis. Therapy may include the use of emollients and keratolytics, such as urea, alpha-hydroxy acids, propylene glycol, salicylic acid, and topical vitamin D analogs and retinoid preparations. Newer synthetic retinoids, such as short- contact topical tazarotene, combined with moisturizers seem promising [10]. Systemic retinoid therapy with acitretin (Soriatane) or isotretinoin (Accutane) can induce dramatic improvement [11]; as it is noticed in one case of the present study. The aim of present report is to publish these cases and for the first time in Iraqi population.
4. Conclusions
- Due to its rarity, EKVP can often go undiagnosed or misdiagnosed for years. So shedding the light on this topic will help the physicians to keep EKVP included in the differential diagnosis when faced with similar cases. This is the first case series reported in Iraq.
DISCLOSURE
- This study was an independent study and not funded by any drug companies.