-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Dermatology and Venereology
2014; 3(5): 84-86
doi:10.5923/j.ajdv.20140305.02
Generalized Hyperpigmentation, in Sudanese Patient with Primary Addison’s Disease as Autoimmune Manifestation of Extra Digestive Helicobacter pylori Infection
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAdil H. H. Bashir1, Gamal O. Elhassan2, 3, Abdel Khalig Muddathir4, Anilkumar Mithani5, Tayseer E. Diab6, Ali Thoulifkar A. Imeer7, Ali B. Sidahmed8, Zuhair A. A. Babikir9, Khalid O. Alfarouk1, Lamya A. M. Elhassan10, Ahmed M. El Hassan1
1Institute of Endemic Diseases, University of Khartoum, Khartoum, Sudan
2Uneizah Pharmacy College, Qassim University, Alqassim, KSA
3Faculty of Pharmacy, Omdurman Islamic University, Khartoum, Sudan
4Faculty of Pharmacy, University of Khartoum, Khartoum, Sudan
5Faculty of Medicine, University of Bahri, Bahri, Sudan
6Faculty of Medicine, Al-Neelain University, Khartoum, Sudan
7ALyrmouk College, Khartoum, Sudan
8AL-Doctors Clinic, Khartoum, Sudan
9University of Medical Sciences and Technology, Khartoum, Sudan
100School of Medicine, Ahfad University for Women, Omdurman, Sudan
Correspondence to: Adil H. H. Bashir, Institute of Endemic Diseases, University of Khartoum, Khartoum, Sudan.
| Email: |  |
Copyright © 2014 Scientific & Academic Publishing. All Rights Reserved.
Addison's disease, or chronic adrenocortical insufficiency, is the overproduction of adrenocorticotropic hormone, ACTH, by the pituitary gland as a compensatory mechanism for decreased cortisol production by the adrenal glands. Classically, patients affected with Addison's disease develop weakness, anorexia, electrolyte imbalances: decreased sodium and chloride with increased serum potassium resulting in hypotension, and hyperpigmentation of the skin and mucous membranes.We report an undiagnosed localized hyperpigmented primary autoimmune Addison’s disease as extra digestive manifestation of Helicobacter pylori infection in a 43 years old male patient, teacher, who presented with localized Palmar dark pigmentation, for 7 months duration. The case was diagnosed and confirmed by ACTH stimulating test and cortisol blood level, and is considered to be the first case (reported in Sudan) associated with H. pylori and it responded to triple therapy.
Keywords: Primary addison's disease, Helicobacter pylori, Autoimmune disease
Cite this paper: Adil H. H. Bashir, Gamal O. Elhassan, Abdel Khalig Muddathir, Anilkumar Mithani, Tayseer E. Diab, Ali Thoulifkar A. Imeer, Ali B. Sidahmed, Zuhair A. A. Babikir, Khalid O. Alfarouk, Lamya A. M. Elhassan, Ahmed M. El Hassan, Generalized Hyperpigmentation, in Sudanese Patient with Primary Addison’s Disease as Autoimmune Manifestation of Extra Digestive Helicobacter pylori Infection, American Journal of Dermatology and Venereology, Vol. 3 No. 5, 2014, pp. 84-86. doi: 10.5923/j.ajdv.20140305.02.
1. Background
- Skin color is highly individual and the variations are controlled by numerous genes. Immunologic or toxic mediated destructions of melanocytes can end in pigmentation disorders. They can manifest locally or diffuse. Generalized hyperpigmentation only rarely reflects a primary genetic disorder but is most often from acquired diseases as in Addison’s disease, secondary hemochromatosis or primary biliary cirrhosis [1]. Neuroendocrine and immune systems recently, as a result of molecular genetic studies provided evidences for common languages of these systems by various signals including neurotransmitters, hormones, and cytokines. It is proven that the immune system is able to produce neurotransmitters and hormones and endocrine organs can even result in cytokines. This new integrative approach allows investigating the physiologic events and diseases as interactions between the psycho-neuro-endocrine-immune systems. The autoimmune polyendocrine syndromes constitute a heterogeneous group of disorders characterized by loss of immune tolerance to self-antigens. In spite of distinct clinical pictures, molecular genetic studies revealed a common molecular mechanism in the associations of organ-specific diseases [2]. Addison disease (primary insufficiency of adrenal cortex) is characterized by clinical signs and symptoms associated with deficiency of adrenal hormones [3].Addison's disease, results in glucocorticoid and mineralocorticoid deficiency. Orthostatic hypotension, fever, and hypoglycemia characterize acute adrenal crisis, whereas chronic primary adrenal insufficiency presents with a more insidious history of malaise, anorexia, diarrhea, weight loss, joint, and back pain. The cutaneous manifestations include darkening of the skin especially in sun-exposed areas and hyperpigmentation of the palmar creases, frictional surfaces, vermilion border, recent scars, genital skin, and oral mucosa. Measurement of basal plasma cortisol is an insensitive screening test. Synthetic adrenocorticotropin 1-24 at a dose of 250µg works well as a dynamic test. Elevated plasma levels of adrenocorticotropin and renin confirm the diagnosis [4].Cutaneous pigmentation is a hallmark of Addison disease. When present, the hyperpigmentation generally localizes to sun-exposed surfaces. Some cases show less well-recognized cutaneous features that is pathognomonic for the disease: oral mucous membrane hyperpigmentation [5].The most frequent etiopathogenesis of Addison disease is related with autoimmunization. In sera of Addison patients there are detectable autoantibodies against other endocrine glands; pituitary microsomal antigens can be frequently detected. The most frequent are antibodies against 55, 60 and 67 kDa antigens [3].Autoimmune Addison's disease is caused by autoreactivity towards the adrenal cortex involving 21-hydroxylase autoantibodies and autoreactive T cells. Autoimmune destruction of the adrenal cortex is triggered by hitherto unknown environmental factors in individuals with genetic susceptibility. Several genes have been identified, of which the major histocompatibility complex haplotypes DR3-DQ2 and DR4-DQ8 are most strongly associated. In addition, other genes also implicated in other autoimmune diseases are linked to Addison's disease, such as cytotoxic T lymphocyte antigen 4 (CTLA-4), protein tyrosine phosphatase non-receptor type 22 (PTPN22), major histocompatibility complex class II transactivator (CIITA), and most recently the C-lectin type gene (CLEC16A). Studies employing T cells in humans and animal models, and the collection of large patient cohorts facilitating genome-wide screening projects, will hopefully improve the understanding of the pathogenesis of the disease in the near future [6] .
2. The Case
- A male patient, 43 years old, married, teacher, descent from non relative parents, resident in Khartoum state.The condition started 7 months ago with insidious onset and progressive course, of localized pigmentation, no weakness, and malaise, loss of appetite or dizziness. Dark pigmentation, localized to the hands was noticed (See Figures 1, 2). Lesions appeared at gums were few mild pigmentation. The Patient did not receive any treatment.General examination: Patient had normal blood pressure and no generalized hyperpigmentation, the general condition was good, not pale, and not icteric. Also, spleen, liver and lymph nodes were not enlarged. There was No ascites or bone involvement.Dermatological examination, Localized hyperpigmented skin, of both palms. Gingiva, proximal dorsal tongue, flexures, pressure sites and palmoplantar creases hyperpigmentation were not involved. No abnormalities were detected in the Face, hair, oral cavity and nails.
 | Figure 1. Shows hyperpigmented palms and palmar creases |
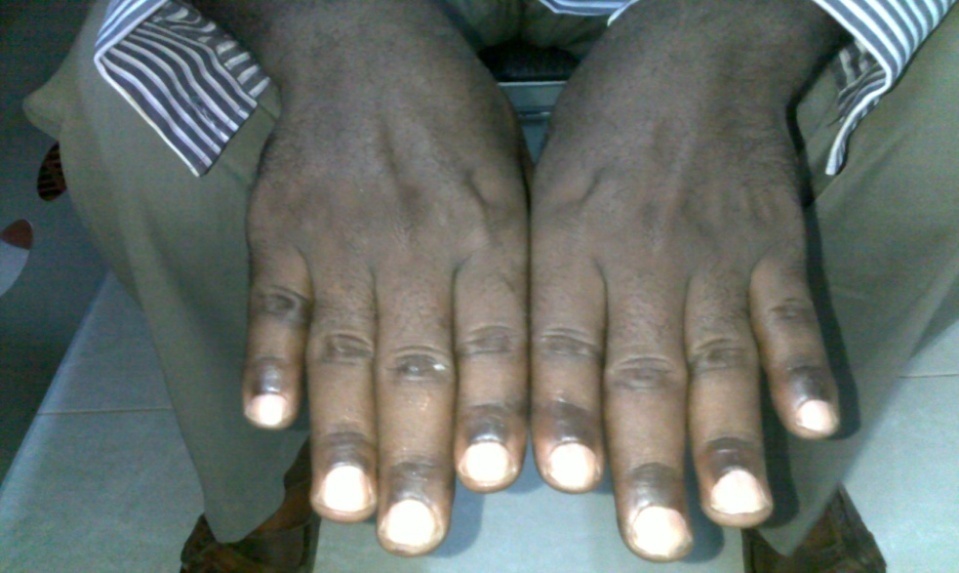 | Figure 2. Shows hyperpigmented nail folds and knuckles of hands dorsi |
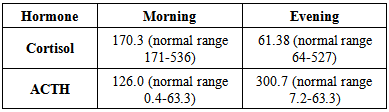 Renal profile:
Renal profile: 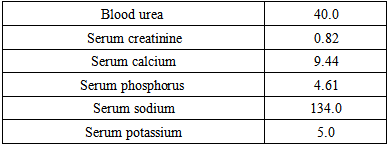 Helicobacter pylori ELISA test: Reactive (25 unit).Helicobacter pylori Ag test: Not detected (4 weeks later).Abdominal U/S: No evidence of suprarenal mass, hyperplasia or atrophy.No evidence of renal parenchymal disease or stone.
Helicobacter pylori ELISA test: Reactive (25 unit).Helicobacter pylori Ag test: Not detected (4 weeks later).Abdominal U/S: No evidence of suprarenal mass, hyperplasia or atrophy.No evidence of renal parenchymal disease or stone. 3. Discussion
- Addison's disease, or chronic adrenocortical insufficiency, is the overproduction of adrenocorticotropic hormone by the pituitary gland as a compensatory mechanism for decreased cortisol production by the adrenal glands. Etiology in this case was probably autoimmune. The case presented with generalized hyperpigmentation, hypotension, abdominal pain, nausea, diarrhea, tiredness and dizziness and, no weight gain for 9 months. On examination, he had hypotension and generalized hyperpigmentation that was more marked on gingival mucosa and frictional sites. Serum cortisol was markedly decreased, while ACTH was remarkably raised and his UltraSonography abdomen showed No adrenal atrophy. On the basis of clinical and laboratory findings, a diagnosis of Addison disease was made. Identification of predisposing genetic helps to understand the common mechanisms and provide possibility for early therapy and prevention as well. In sera of this case of Addison patient’s autoantibodies against pituitary microsomal antigens can be frequently detected as seen in H. pylori ELISA test show reactive and considered as autoantibodies where triple therapy given to show good clinical and investigatory response to show first case of H. pylori extra digestive manifestation. Treatment of pigmentation disorders are based on a diagnosis which sometimes allows a specific intervention, although cosmetically acceptable results are difficult to obtain. Here in this case report, we focus on the subtle findings of diffuse hyperpigmentation and intermittent but repetitive chronic dyspepsia symptoms to correctly identify the diagnosis of Addison's disease effectively and efficiently.
ACKNOWLEDGMENTS
- This work has been supported by Alfarouk Biomedical Research LLC.