-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Condensed Matter Physics
p-ISSN: 2163-1115 e-ISSN: 2163-1123
2024; 13(3): 45-50
doi:10.5923/j.ajcmp.20241303.01
Received: Dec. 2, 2024; Accepted: Dec. 23, 2024; Published: Dec. 28, 2024

Ab Initio Study of Hydrogen Adsorption on Lanthanum Chromium Oxide (LaCrO3) (001) Surface as Potential Solid Oxide Fuel Cell Material
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLOyedare Peter Olusola1, Jibril Ibrahim Manya2, Maharaz M. Nasir3, Alhassan Shuaibu2
1Science Laboratory Department Federal Polytechnic Ede, Osun State, Nigeria
2Department of Physics, Kaduna State University, Kaduna, Kaduna, Nigeria
3Department of Physics Federal University Dutse, Jigawa State
Correspondence to: Alhassan Shuaibu, Department of Physics, Kaduna State University, Kaduna, Kaduna, Nigeria.
| Email: |  |
Copyright © 2024 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Density functional theory plus Hubbard (DFT+U) calculations were employed to investigate the adsorption of Hydrogen (H)-adatom on the (001) surface of LaCrO3 (LCrO). The adsorption is found to be stable with H binding preferentially at Cr site on the LaO-terminated surface. The adsorption of H molecule also leads to the electrons transferring from the substrate to the charges rearrangement within the compound. We further predict the adsorption energies of Hydrogen adsorption sites on LCrO (001) surface. Based on the adsorption energy comparison, LCrO is more hydrogenation-tolerant than traditional Ni-based anode materials, which is qualitatively in line with available experimental results. This study provides a scientific basis for rational design of Hydrogen-tolerant intermediate temperature materials for Solid oxide fuel cell (SOFCs).
Keywords: Density functional theory plus Hubbard (DFT+U), LaCrO3 (LCrO), Hydrogen adsorption and Solid oxide fuel cell (SOFC)
Cite this paper: Oyedare Peter Olusola, Jibril Ibrahim Manya, Maharaz M. Nasir, Alhassan Shuaibu, Ab Initio Study of Hydrogen Adsorption on Lanthanum Chromium Oxide (LaCrO3) (001) Surface as Potential Solid Oxide Fuel Cell Material, American Journal of Condensed Matter Physics, Vol. 13 No. 3, 2024, pp. 45-50. doi: 10.5923/j.ajcmp.20241303.01.
1. Introduction
- Solid oxide fuel cell (SOFC) provides a new and clean electric power generation system [1]. At present, Y2O3 stabilized ZrO2 (YSZ) is commonly used for the electrolyte of solid oxide fuel cell [2]. Since the oxide ion conductivity of YSZ is insufficient for the electrolyte of fuel cells, a thin electrolyte film without gas leakage and an excessively high operating temperature such as 1273 K, are essential for the high power density of SOFCs when YSZ is used as the electrolyte [3]. On the other hand, all advantages of SOFC such as a high efficiency and a variety of usable fuel can be obtained at decreased temperatures such as 1073 K. Furthermore, the choice of materials for the cell stacking becomes wide; in particular, cheap refractory metals such as stainless steel will be usable by decreasing the operating temperature down to 1100 K [4]. Consequently, the decrease in the operating temperature becomes the critically important subject for the development of cheap but reliable cells [5]. Decreasing the operating temperature requires an active electrode, in particular, a cathode catalyst and an electrolyte with a low resistance. Ceria doped with Gd or Sm is generally considered for the electrolyte of SOFC operable in the decreased temperature range [6]. However, ceria based oxide exhibits n-type semi conduction in the reducing atmosphere [7]. Consequently, n-type semi conduction drastically decreases the open circuit potential compared to the theoretical values [8]. In addition, some amount of fuel is consumed by oxygen leakage W to the internal short circuited state of the electrolytes by formed electrons. It is also reported that the expansion due to the reduction causes a severe stress on electrolytes which sometimes become higher than the intrinsic mechanical strength of CeO2 fuel cell oxide [9]. Therefore, there are some problems which should be solved for CeO based oxide cells. On the other hand, preparing very thin YSZ film is also investigated for intermediate temperature SOFC [10], however, the reliability becomes low when the thickness of the electrolyte becomes extremely thin and furthermore, it is anticipated that the power density may become unstable by using very thin YSZ film for electrolyte.It is, therefore, of great importance to develop new electrolyte materials which exhibit high oxide ion conduction over a wide oxygen partial pressure range. The reports on the oxide ion conductivity are limited on oxides with non-fluorite structure. In a recent study, it was observed that oxide ion conductivity in the oxide with perovskite structure was investigated within both theory and experiment was found that the LaCrO3-based perovskite type oxide exhibits high oxide ion conductivity [11], which is comparable with that of CeOz- based oxide. In particular, LaCrO3 doped with Sr for La and Mg for Cr sites exhibits high oxide ion conductivity stable over a wide oxygen partial pressure range, the high oxide ion conductivity of this oxide system was reported by several other researchers. The advantage of this oxide is that almost pure oxide ion conductivity is exhibited in both reducing and oxidizing atmospheres. It is reported that the electron and hole conduction is smaller than that of oxide ion by a few orders of magnitude. It is, therefore, expected that the operating temperature of SOFC can be decreased by using LaCrO3-based oxide for the electrolyte of SOFCs. But a major drawback of LaCrO3-based perovskite when to be consider as a solid oxide fuel cell (SOFC) material is that it can experience structural and performance instability. Another issue to be considering is that the cathode the site of the oxygen reduction reaction becomes the limiting component in a SOFC as temperatures are lowered, when LaCrO3 is to be used. Hence, by surface modification, or doping such drawback can be address. The Hydrogen (H) absorption on surface of other perovskite material has shown such ability, therefore, this work used computational method to study the hydrogen Absorption on LaCrO3 surface within the frame of Density functional theory plus Hubbard (DFT+U) As Potential Material for Applications in Intermediate Temperature Solid Oxide Fuel Cells.
2. Computational Details
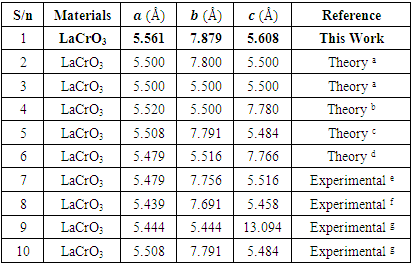
- First-principles DFT+U total energy calculation are carried out as implemented in the Quantum ESPRESSO code [12], in which the plane-wave pseudopotential total energy calculation method based on the DFT is used. The interaction between nuclei and electrons is approximated with Vanderbilt ultrasoft pseudopotential [13] and the Perdew-Wang-91 parametrization [14] is taken as the exchange-correlation functional in the generalized- gradient approximation. After test calculation, kinetic energy cutoff at 420 eV and the Brillouin zone sampling 12×12×1 and 8×8×1 Monkhorst–Pack [15] k-points for surface is adopted. By further increasing the kinetic energy cutoff and the number of k-points the change in the results could be neglected. La, Cr, O, S, H atoms are described by La(5s25p65d16s2), Cr(3d54s1), O(2s22p4), S(3s23p4), H(1s1) valence electrons, respectively. A relaxation is performed for the constructed supercell by using Broyden–Fletcher–Goldfarb–Shanno algorithm [16] to minimize the energy with respect to atomic position. In calculations the tolerances for self-consistence are set at 1.0 × 10–6 eV atom–1 for total energy, 0.05 eV Å–1 for force, 2.0 × 10–5 eV atom–1 for band energy, 0.1 GPa for maximum stress and 0.002 Å for the maximum displacement. In this study, we consider orthorhombic perovskite structure of orthorhombic structure with space group Pbnm since the structural phase transition to orthorhombic symmetry was suggested to occur above high temperature [17]. The Jahn–Teller distortion may not be critical under SOFCs operating conditions.We model the adsorbate adsorption on a much less polar LCrO3 (001) surface with respect to LCrO3 (110) surface consisting of alternating CrO2/LaO plane. The structure with GGA+U parameters is shown in Figure 1.
 | Figure 1. Structure of Pure LaCrO3 with GGA+U optimized parameters |
|
|
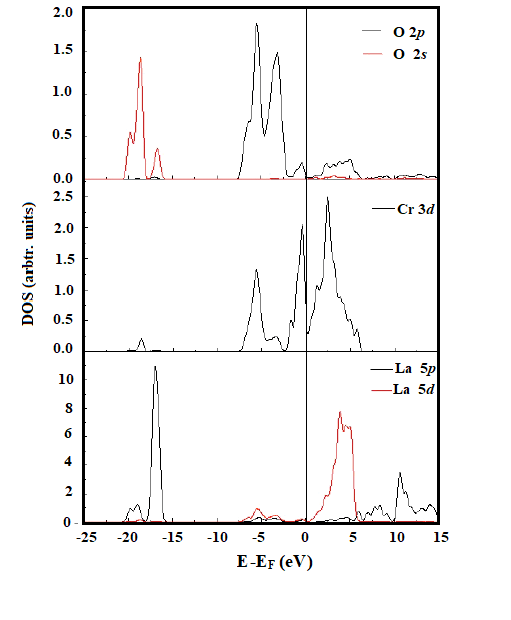 | Figure 2. Calculated PDOS for pure orthorhombic LaCrO3 with GGA+ U optimized parameters. The zero point of the energy axis corresponds to the Fermi level |
|
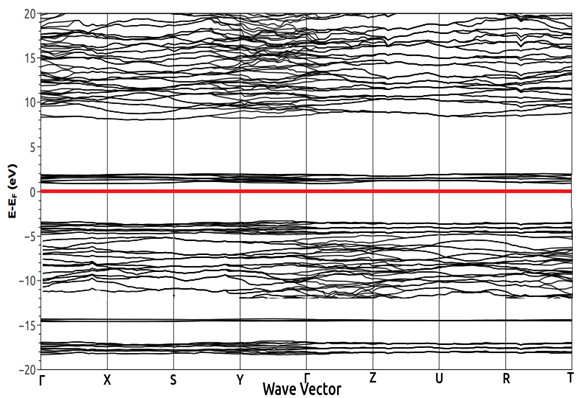 | Figure 3. Calculated Band Structure for Pure LaCrO3 with our optimized GGA+U (U=3.12 eV) parameters (the red line at 0 indicate the Fermi level) |
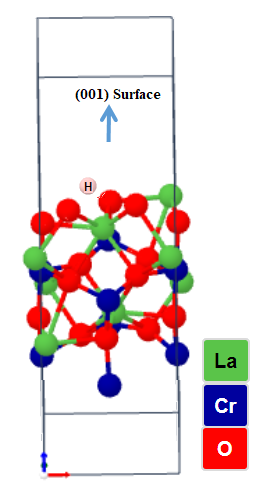 | Figure 4. Schematic representation of the optimized LCrO3 (001) surface with Hydrogen terminated adatom |
|
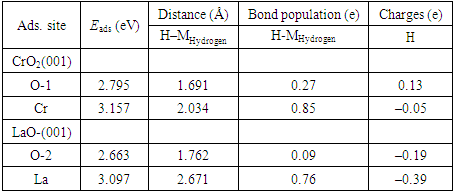
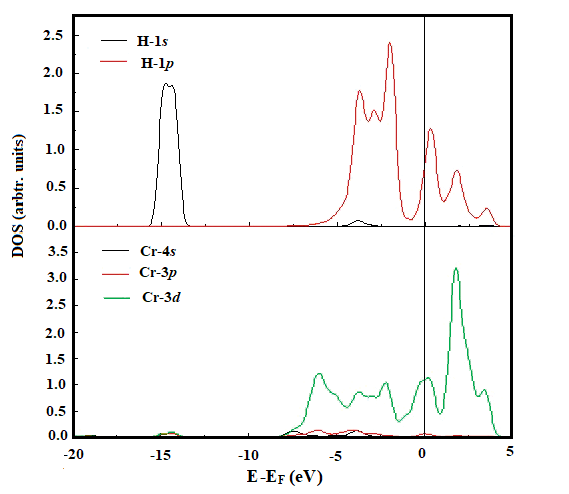 | Figure 5. The PDOS for adatom H in Cr sites and the nearest neighbor Cr atom on the surface. The Fermi level is set to zero on the energy scale |
3. Conclusions
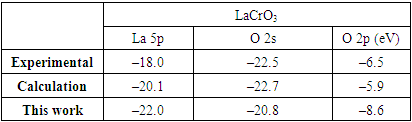
- Hydrogen tolerance on the perovskite-type materials LCrO3 is investigated by means of DFT+U calculations. Our calculated PDOS of bulk LCrO3 shows good agreement with the photoemission data and theoretically calculated values. Hydrogen adsorption is found to be stable with binding preferentially at Cr site on the LaO-terminated surface. The adsorption of Hydrogen adatom leads to the electrons transferring from the substrate to the adatom and the charges rearrangement within the molecule. In addition, the adsorption results of the corresponding H-containing dissociated species are obtained. Both the species are found to be preferentially adsorbed at the Cr site. Both bond population and PDOS analysis show that there is hybridization between adatom H adatom and surface Cr atoms. Based on the adsorption energy comparison, LCrO is more Hydrogen-tolerant than traditional Ni-based anode materials used in SOFC, which is qualitatively in line with available experimental results. This study provides a meaningful basis for hydrogen-tolerant anode development as intermediate temperature in SOFC.
ACKNOWLEDGEMENTS
- We thank and acknowledged the Department of Physics, Kaduna State University for providing the computing facilities used in this work.