-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Condensed Matter Physics
p-ISSN: 2163-1115 e-ISSN: 2163-1123
2024; 13(2): 35-44
doi:10.5923/j.ajcmp.20241302.02
Received: Jun. 26, 2024; Accepted: Jul. 22, 2024; Published: Jul. 27, 2024

Effect of Halogens on the Structural and Electronic Properties of Diphenyl-Diketopyrrolopyrrole and Its Derivatives
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLHindreen A. Ibrahim1, Salah M. A. Ridha2, Najlaa Ozaar Hasan3
1Basic Sciences Branch, College of Dentistry, University of Kirkuk, Iraq
2Department of Physics, College of Science, University of Kirkuk, Iraq
3Department of Physics, College of Girls Education, University of Kirkuk, Iraq
Correspondence to: Hindreen A. Ibrahim, Basic Sciences Branch, College of Dentistry, University of Kirkuk, Iraq.
| Email: |  |
Copyright © 2024 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

In the present study, Structural, UV spectroscopic properties and quantum chemical calculations have been performed on theoretical study of the diphenyl-diketopyrrolopyrrole with its derivatives (chlorodiphenyl-diketopyrrolopyrrole, bromodiphenyl-diketopyrrolopyrrole and fluorodiphenyl-diketopyrrolopyrrole) were studied. Using the DFT/B3LYP/6-311G (d,p) level of theory in the gas phase, the compounds' ground state geometries have been optimised. The assigned chemical structure of molecules was confirmed using IR spectroscopic technique. The UV-Vis spectral properties, maximum wavelength, energy and oscillator strength of three compounds were predicted by the TD-DFT approach. The extracted molecular structure has served as the basis for geometric optimisations. The structural and geometric characteristics were computed, and theoretical results were compared to experimental X-ray values obtained from the literature. The computationally calculated structural and geometric characteristics correlate well with the experimentally acquired x-ray values that documented from literature. The effects of the halogen substituent on the characteristic diphenyl-diketopyrrolopyrrole bands in the UV-visible spectral are discussed. In addition, Chemical reactivity descriptors on a global scale, such as frontier molecular orbital analysis, were used to the compounds' optimal structures in order to address their reactive qualities.
Keywords: Diphenyl-diketopyrrolopyrrole, DPPs, DFT, Geometry, Energy gap, Global reactivity descriptors
Cite this paper: Hindreen A. Ibrahim, Salah M. A. Ridha, Najlaa Ozaar Hasan, Effect of Halogens on the Structural and Electronic Properties of Diphenyl-Diketopyrrolopyrrole and Its Derivatives, American Journal of Condensed Matter Physics, Vol. 13 No. 2, 2024, pp. 35-44. doi: 10.5923/j.ajcmp.20241302.02.
Article Outline
1. Introduction
- The shortage of fossil fuels and the increasing demand for energy have prompted people to develop new types of clean and sustainable energy [1] [2]. In recent times, researchers have observed significant advancements in organic semiconductors, which provide numerous advantages such as flexibility, reduced cost, facile synthesis, scalability, and decreased weight [1] [3] [4]. Diketopyrrolopyrrole (DPP), out of all the organic semiconductors that have been documented, has demonstrated remarkable efficacy as a fundamental component in the fabrication of high-performance functional materials applicable to a diverse array of electronic devices [5]. Diketopyrrolopyrrole (DPP) discovered in 1974 by Farnum et al [6], and had not been extensively utilized until Iqbal and his colleagues put forth a comparatively rational synthetic mechanism, until after 10 later. Subsequent to its commercial introduction in 1986 under the name C.I. Pigment Red 255, diphenyl-DPP was rapidly followed by a succession of analogs featuring various substitutes (henceforth referred to as DPPs), which amassed substantial market shares in the pigment industry [7]. Subsequently, DPP gained extensive use as a pigment in inks, as well as in paints for the purpose of coating. Although DPP-based conjugated compounds were first described in 1993, their application as semiconductors in organic electronics did not become widespread until 2008. This was the year that Nguyen, Janssen, and Winnewisser successfully synthesized DPP-based semiconductors that could be processed in solution for usage in OFETs and OPVs. Since 2008, Diketopyrrolopyrrole (DPP) has been acknowledged as a very promising conjugated building block for the fabrication of high-performance semiconducting materials used in a wide range of electronic devices [8]. The planar structure of an organic dye has been demonstrated to make it a promising option for photothermal therapy (PTT) due to its favourable photostability, strong fluorescence, exceptional molar absorption coefficient, and convenient modifiability [9], possesses exhibits distinctive characteristics, including excellent conjugation, strong electron-drawing capability, thermal and photostability stability, and a high quantum efficiency for fluorescence [10]. In addition to its low-bandgap characteristics and the ability to tune energy levels appropriately, the fused highly planar Diketopyrrolopyrrole (DPP) unit is capable of facilitating considerable intermolecular π–π interactions, which in turn improves the mobility of charge carriers [11]. The incorporation of different monomeric units (either donor or acceptor) with DPP enables precise manipulation of the energy levels of (HOMO) the highest occupied molecular orbital and lowest unoccupied molecular orbital (LUMO). By manipulating these factors, the hole mobility of p-type materials has surpassed
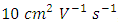 , whereas the electron mobility of n-type semiconductors has achieved over
, whereas the electron mobility of n-type semiconductors has achieved over 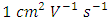 [5]. The aromatic ring substituents play an important function in the conjugate system, influencing the maximum absorption and emission wavelengths of the entire molecule [12]. Due to their high electron mobility, DPPs have garnered significant attention in the field of energy, leading to increased interest [8]. From their accidental synthesis in the 1970s to their subsequent widespread use as a high-quality pigment twenty years later, and now to their current status as a promising new energy solution, Diketopyrrolopyrrole (DPPs) have been blessed with ever-evolving values and concepts. The energy area has received a lot of attention and resources thus far [7]. However, the primary purpose of DPPs, which is to enhance our lives with vibrant hues, is being disregarded. The chromophore of DPPs consists of a central core composed of two fused pyrrolidone rings, as indicated by its name. The most basic derivative of this kind, diphenyl-DPP, has a maximum absorption at 538 nm in solid state, resulting in a red color. Other derivatives within the DPP family can exhibit either a violet or orange color variation, depending on whether their absorption is shifted towards the blue or red end of the spectrum. This shift is caused by the electron donating or withdrawing impact of conjugation. The distinctive characteristics of DPPs mostly arise from the π-π stacking interactions between aromatic rings and the hydrogen bonding facilitated by lactam units. DPPs possess a robust intermolecular connection that grants them an exceptionally elevated lattice energy [7]. Because to their strong fluorescence properties and exceptional stability [1]. Small molecules derived from DPP have found extensive use in a variety of organic electronic devices over the past few decades [9]. These devices include sensing and bio-imaging [13], organic field-effect transistors [14], organic solar cells [15], ambipolar transistors, dye-sensitized organic solar cells [16], and bulk heterojunction solar cells (BHJ) [17], organic light emitting diodes (OLEDs) [16], memory devices, photodetectors [5], organic light emitting diodes [18], organic photovoltaics (OPVs) [19], fluorescent sensors [20], and optical imaging contrast agents, Fluorescent probes based on DPP have been created for a range of analytes [20] including thiols, reactive oxygen species, anions, cations, pH, H2, and CO2 [21], two-photon absorption [22], gas detectors, solid-state dye lasers, [23], and chemical sensors [24], Plus, DPP derivatives' unique characteristics have led to their remarkable PDT and PTT efficacy in cancer therapy [20], partly due to the synthetic capabilities that allow for the modification of their molecular characteristics to create materials with unique functionalities. Originally, Ciba-Geigy created their synthesis and made the compounds available for sale as automotive pigments [13]. High-performance pigments derived from DPPs are widely used [25]. The compounds have vivid colours (varying from yellow-orange to red-violet) and are extremely resistant to light, heat, chemical, and climate impacts [23]. Additionally, in normal pigment molecule terms, their very low Mr makes certain of their physical features, such high melting temperatures, unusual. Incorporating (DPP) units into different materials, such as polymers, has been found to have beneficial effects [26], polymer–surfactant complexes, oligomers and dendrimers [25], resulting in intensely variegated and photoluminescent [27], and electroluminescent materials [28]. Solubility is a crucial factor in the fabrication of any device, whether it is in form of thin film consisting of Diketopyrrolopyrrole (DPP) or involves the integration of the Diketopyrrolopyrrole (DPP) unit into a supporting matrix. Diketopyrrolopyrrole (DPP) compounds exhibit limited solubility in most commonly used solvents. Although this characteristic is advantageous for some applications, the ability to dissolve these compounds would enable to use of solution-based methods such as drop-casting, spin-coating, and inkjet printing for the fabrication of DPP-based devices [25]. In the present study, the Optimized geometry, Optical properties and Chemical Description of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) orbital positions required for effective charge transfer, etc, of diphenyl-diketopyrrolopyrrole and its derivatives using density functional theory (DFT) method Hybrid three-parameters Lee–Yang–Par (B3LYP) exchange correlation in combination with 6-311G(d,p) basis set have been studied. None of the compounds has been studied experimentally, but the experimental data of DPP without phenyl groups was obtained [29]. The occurrence of quantum chemical investigations on these compounds is notably infrequent, the obtained results are discussed and compared with similar literatures of the studied molecules.
[5]. The aromatic ring substituents play an important function in the conjugate system, influencing the maximum absorption and emission wavelengths of the entire molecule [12]. Due to their high electron mobility, DPPs have garnered significant attention in the field of energy, leading to increased interest [8]. From their accidental synthesis in the 1970s to their subsequent widespread use as a high-quality pigment twenty years later, and now to their current status as a promising new energy solution, Diketopyrrolopyrrole (DPPs) have been blessed with ever-evolving values and concepts. The energy area has received a lot of attention and resources thus far [7]. However, the primary purpose of DPPs, which is to enhance our lives with vibrant hues, is being disregarded. The chromophore of DPPs consists of a central core composed of two fused pyrrolidone rings, as indicated by its name. The most basic derivative of this kind, diphenyl-DPP, has a maximum absorption at 538 nm in solid state, resulting in a red color. Other derivatives within the DPP family can exhibit either a violet or orange color variation, depending on whether their absorption is shifted towards the blue or red end of the spectrum. This shift is caused by the electron donating or withdrawing impact of conjugation. The distinctive characteristics of DPPs mostly arise from the π-π stacking interactions between aromatic rings and the hydrogen bonding facilitated by lactam units. DPPs possess a robust intermolecular connection that grants them an exceptionally elevated lattice energy [7]. Because to their strong fluorescence properties and exceptional stability [1]. Small molecules derived from DPP have found extensive use in a variety of organic electronic devices over the past few decades [9]. These devices include sensing and bio-imaging [13], organic field-effect transistors [14], organic solar cells [15], ambipolar transistors, dye-sensitized organic solar cells [16], and bulk heterojunction solar cells (BHJ) [17], organic light emitting diodes (OLEDs) [16], memory devices, photodetectors [5], organic light emitting diodes [18], organic photovoltaics (OPVs) [19], fluorescent sensors [20], and optical imaging contrast agents, Fluorescent probes based on DPP have been created for a range of analytes [20] including thiols, reactive oxygen species, anions, cations, pH, H2, and CO2 [21], two-photon absorption [22], gas detectors, solid-state dye lasers, [23], and chemical sensors [24], Plus, DPP derivatives' unique characteristics have led to their remarkable PDT and PTT efficacy in cancer therapy [20], partly due to the synthetic capabilities that allow for the modification of their molecular characteristics to create materials with unique functionalities. Originally, Ciba-Geigy created their synthesis and made the compounds available for sale as automotive pigments [13]. High-performance pigments derived from DPPs are widely used [25]. The compounds have vivid colours (varying from yellow-orange to red-violet) and are extremely resistant to light, heat, chemical, and climate impacts [23]. Additionally, in normal pigment molecule terms, their very low Mr makes certain of their physical features, such high melting temperatures, unusual. Incorporating (DPP) units into different materials, such as polymers, has been found to have beneficial effects [26], polymer–surfactant complexes, oligomers and dendrimers [25], resulting in intensely variegated and photoluminescent [27], and electroluminescent materials [28]. Solubility is a crucial factor in the fabrication of any device, whether it is in form of thin film consisting of Diketopyrrolopyrrole (DPP) or involves the integration of the Diketopyrrolopyrrole (DPP) unit into a supporting matrix. Diketopyrrolopyrrole (DPP) compounds exhibit limited solubility in most commonly used solvents. Although this characteristic is advantageous for some applications, the ability to dissolve these compounds would enable to use of solution-based methods such as drop-casting, spin-coating, and inkjet printing for the fabrication of DPP-based devices [25]. In the present study, the Optimized geometry, Optical properties and Chemical Description of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) orbital positions required for effective charge transfer, etc, of diphenyl-diketopyrrolopyrrole and its derivatives using density functional theory (DFT) method Hybrid three-parameters Lee–Yang–Par (B3LYP) exchange correlation in combination with 6-311G(d,p) basis set have been studied. None of the compounds has been studied experimentally, but the experimental data of DPP without phenyl groups was obtained [29]. The occurrence of quantum chemical investigations on these compounds is notably infrequent, the obtained results are discussed and compared with similar literatures of the studied molecules.2. Results and Discussions
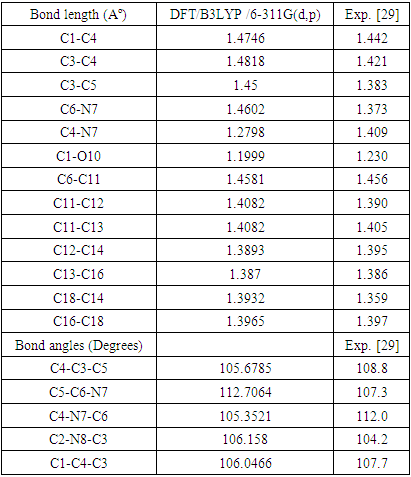
2.1. Optimized Geometry
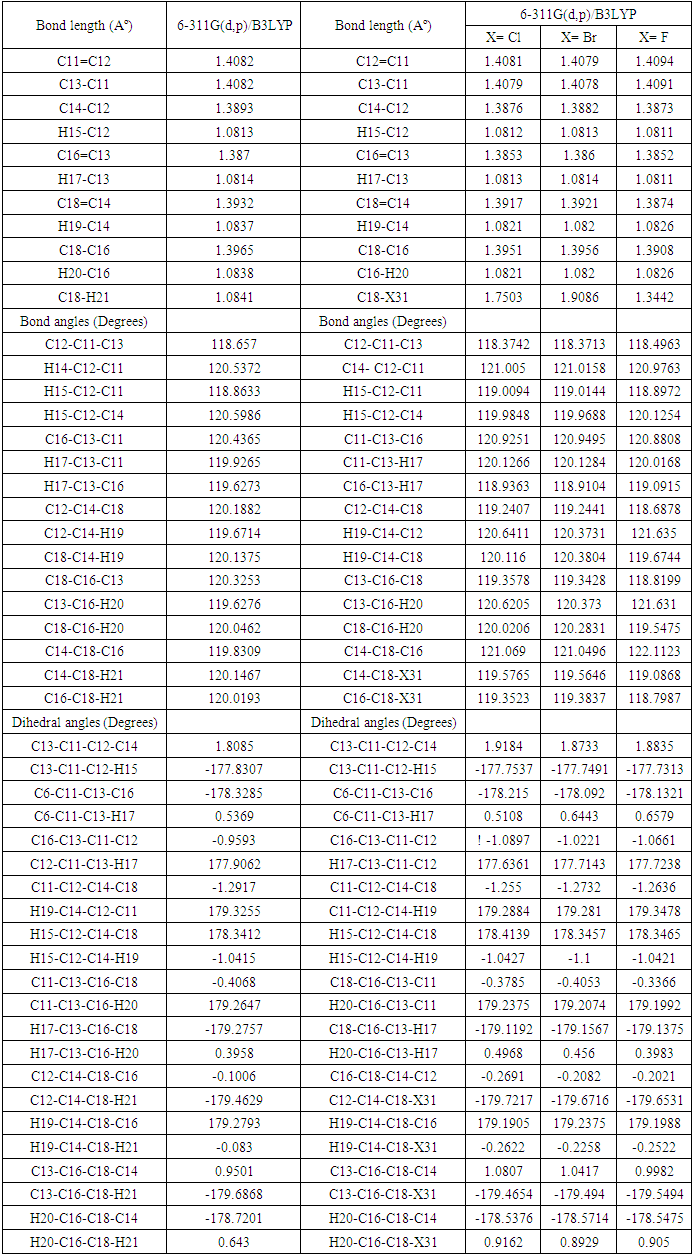
- The geometry optimized of all four compounds based on quantum chemical computations using density functional theory (DFT), hybrid three-parameters B3LYP exchange correlation in combination with 6-311G(d,p) basis set. No instances of imaginary frequencies were observed following the diagonalization of the Hessian matrix, so validating the authenticity of the computed geometries as genuine minima on the hypersurfaces of the ground state. The confirmation of these minima as absolute minima in relation to phenyl rotations was obtained through the optimisations conducted using differently rotated initial geometries. The calculations for the single point energy of the ground state were performed on the geometries obtained from the optimization technique. Only for (DPP) are available experimental X-ray structures [29]. The essential calculated bond lengths and valence angles are presented in Tables 1 and 2. The computed geometric analysis of diphenyl-diketopyrrolopyrrole and its derivatives accurately predicts consistent bond lengths in phenyl rings and alternating bond lengths in heterocycles. Nevertheless, it is unfeasible to quantitatively measure the disparities between the calculated bond lengths and the experimental values using the literature [29]. In fact, the computed values differ more than 0.03 A°. In general, the computed geometry exhibits a higher degree of proximity to SAPDES01 [29]. The two benzene rings are regular hexagons with bond lengths that fall between that of a single (1.3958) and double (1.3961) bond. This is consistent with the usual values reported in the literature for one bond (1.54) and two bonds (1.33) [30], since the bond lengths for the two varied (0.144) for single and (0.066) for double, and the C-H bond lengths between 1.0827 and 1.0841 Å is in agreement with [31] between 1.0853 and 1.0870Å, as the differ 0.0026-0.0029Å. The interatomic distance within the diketopyrrolopyrrole ring are not equal the average value for C=N bond lengths of (DFT) is 1.28Å, and these values show the C=N bond lengths average value significantly longer than the normal C=N double bond (1.22Å) [32]. The C1=O10 bond lengths is 1.1999Å (DFT) and the experimental 1.230Å [29] has the differ more than 0.03Å. Figure 2(a) illustrates the optimised molecular structure of diphenyl-diketopyrrolopyrrole derivatives, with atom numbering provided. Figure 2(b) displays the crystal structure produced for these compounds, wherein X represents chlorine, bromine, or fluorine. The optimised structural parameters of chloro, bromo, and fluoro-diphenyl-diketopyrrolopyrrole were determined using the density functional theory (DFT) method using the B3LYP functional and the 6-311G(d,p) basis set. The atom numbering scheme provided in Figure 2 (a) was used during the calculations.
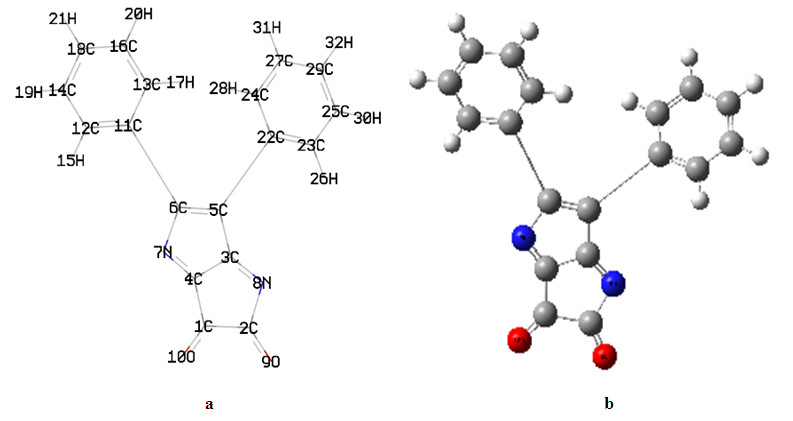 | Figure 1. (a) The atoms numbering and the theoretical geometry of the diphenyl-diketopyrrolopyrrole. (b) Compounds' molecular geometry with atom numbering where (gray, white, blue, red and black is the carbon C, hydrogen H, nitrogen N and oxygen O) respectively |
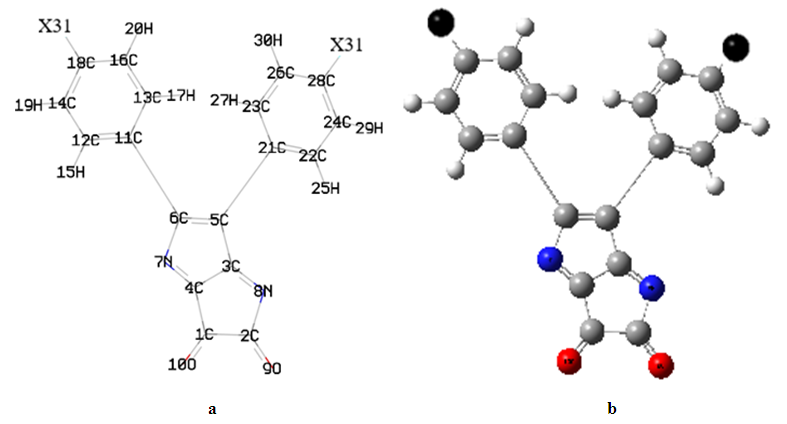 | Figure 2. (a) The atoms numbering and the theoretical geometry of the diphenyl-diketopyrrolopyrrole derivatives. (b) Compounds' molecular geometry with atom numbering where (gray, white, blue, red and black is the carbon C, hydrogen H, nitrogen N, oxygen O and X= chlorine Cl and bromine, Br, fluorine F) respectively |
|
|
2.2. Optical and Electrochemical Properties
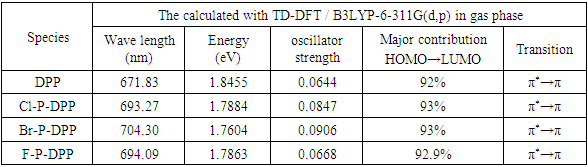
- The Time-dependent (TD-DFT/B3LYP) approach was used to determine the entire UV–VIS absorption spectra of the compounds under study at the B3LYP optimised geometry. Recently, time-dependent density functional approaches have emerged as a useful and reasonably reliable tool for single point electronic excitation computations in a variety of molecular systems, specifically conjugated ones [37]. The optical absorbance of all compounds was evaluated in the gas phase, as shown in Figure 3. The estimation of the optical band gap determined from the absorption spectra are 1.8455, 1.7884, 1.7604 and 1.7863eV for DPP, p-Cl DPP p-Br DPP and p-F DPP respectively. The spectral properties of the diphenyl-diketopyrrolopyrrole and its derivatives are compared in Figure 3 the diphenyl-diketopyrrolopyrrole absorption bands range between 500 nm to 850 nm, Cl-DPP showed absorption band in the visible region from 500–900nm, Br-DPP absorption band in the range 500-900nm, F-DPP absorption band in the visible region from 500–900nm. The absorption spectra show red shifted about 50 nm when comparing diphenyl-DPPs with its derivatives. The semiconducting properties of DPP, Cl-P-DPP, Br-P-DPP, and F-P-DPP in gas phase are shown theoretically in Table 3.
|
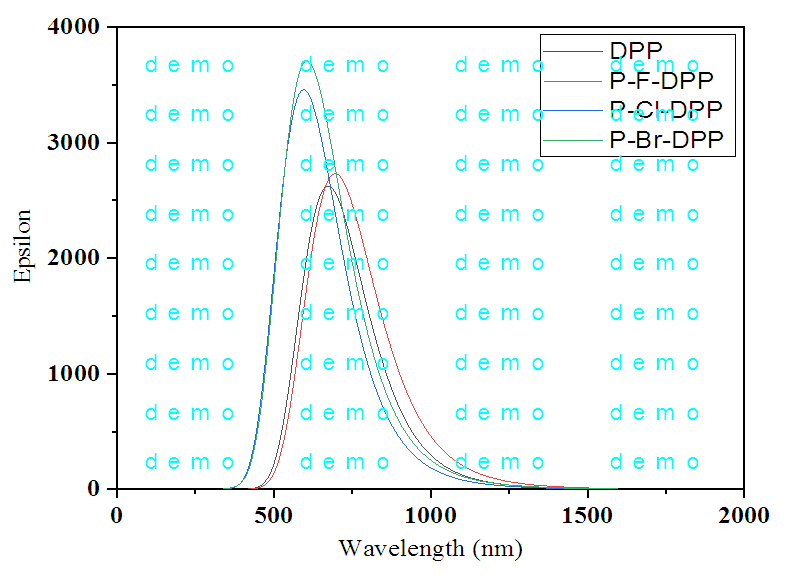 | Figure 3. UV- UV-VIS absorption spectra of DPP and its derivatives |
2.3. Frontier Molecular Orbitals (FMO) Analysis
- Descriptors of global reactivity were analysed to comprehend the connection between global chemical reactivity, structure and stability [38]. The frontier molecular orbitals (FMO) called highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). The eigenvalues of HOMO and LUMO, as well their energy gap, represents the molecule biological activity. A molecule with a short FMO's gap (ΔΕLUMO-HOMO) is more polarisable and has strong chemical reactivity but a low kinetic stability, and vice versa [39]. The ionisation potential is proportional to energy of (HOMO), that can be considered as the outer orbital housing electrons. However, the LUMO is electron-accepting, and the LUMO energy proportional to the electron affinity [39]. FMOs have significant roles in quantum chemistry, optical, and electrical properties [40]. Figure 4 displays the 3D representations of specific molecular orbitals and the corresponding energy gap for each electronic transition. The (HOMO) exhibits localization throughout the entire molecule, whereas the (LUMO) is primarily localised over the diketopyrrolopyrrole and a limited number of phenyl rings. The disparity existing between these entities corresponds to the quantum energy necessary for the stimulation of an electron's transition from (HUMO) to (LUMO), and is equivalent to chemical hardness [38]. The determination of the (HOMO and LUMO) of compounds serves as a valuable tool for predicting the likelihood of noncovalent interactions within those compounds. Ideally, (HOMO) and the (LUMO) should originate from distinct molecules to ensure a favourable interaction [41]. The ionisation energy and electron affinity may be calculated from the HOMO and LUMO orbital energies as: I = -EHOMO, A = -ELUMO [38]. For the DPP, has a low value of EHOMO = -6.6eV, ELUMO = -4.2eV than the Cl-P-DPP, Br-P-DPP and F-P-DPP that have EHOMO = -6.77, -6.7 and -6.68eV, ELUMO = -4.47, -4.46 and -4.37eV, energy gap = ELUMO - EHOMO = 1.8455 eV more than the Cl-P-DPP, Br-P-DPP and F-P-DPP are 1.7884, 1.7604 and 1.7863 eV, ionization potential I = 6.6eV and electron affinity A = 4.2eV, has a value less than the Cl-P-DPP, Br-P-DPP and F-P-DPP that have ionization potential I = 6.77, 6.7 and 6.68eV, electron affinity A = 4.47, 4.46 and 4.37eV, respectively.
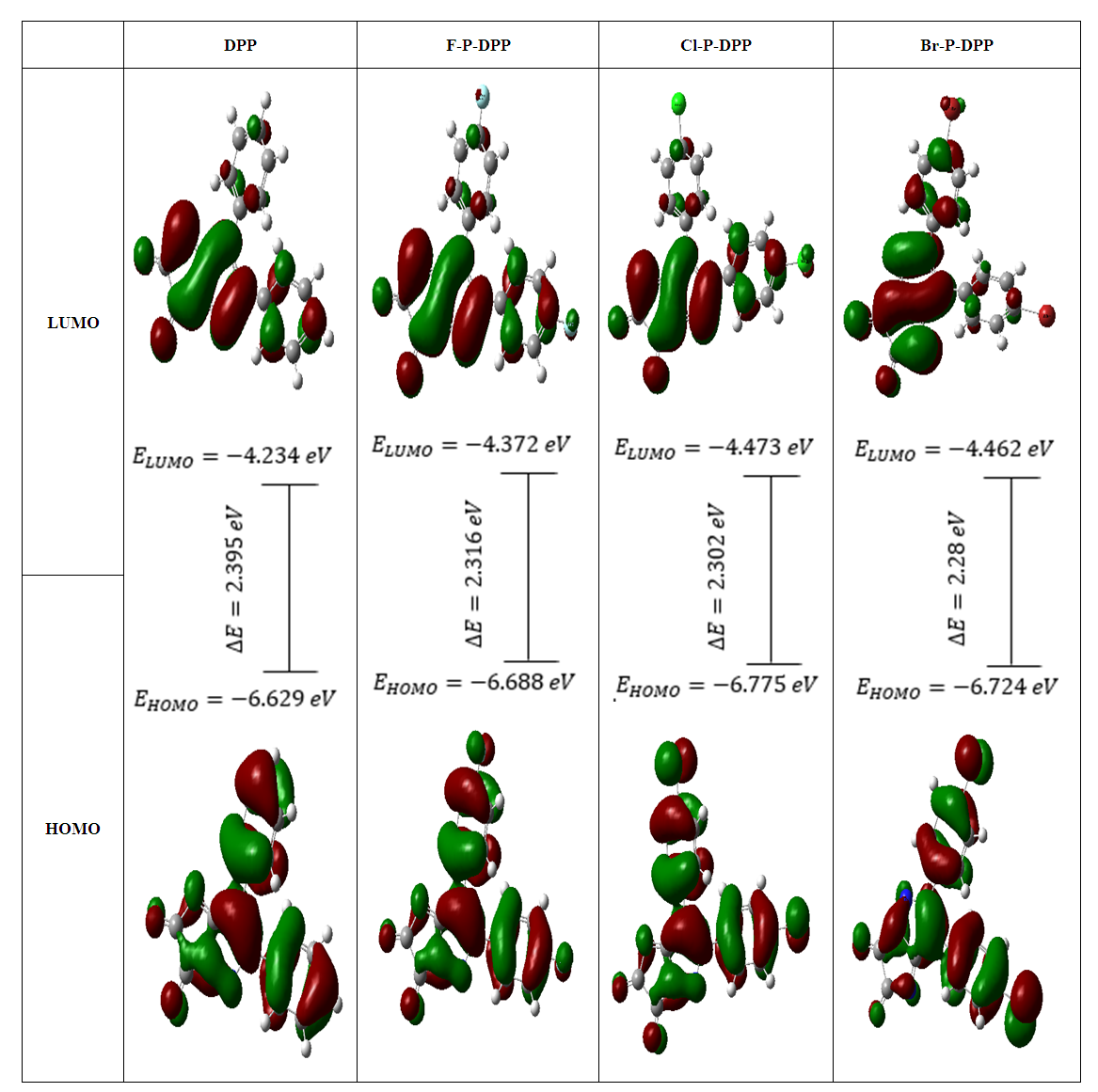 | Figure 4. Molecular orbitals energies |
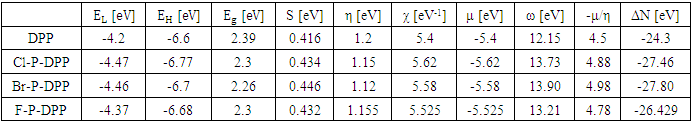
 | (1) |
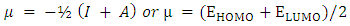 | (2) |
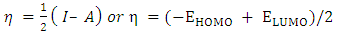 | (3) |
 | (4) |
 | (5) |
 | (6) |
3. Conclusions
- In the present study, the molecular structure and UV-visible spectral of diphenyl-diketopyrrolopyrrole with its derivatives (chlorodiphenyl-diketopyrrolopyrrole, bromodiphenyl-diketopyrrolopyrrole and fluorodiphenyl-diketopyrrolopyrrole) have been calculated using the DFT/B3LYP/6-311G (d,p) level of theory in gas phase. The theoretical results that were available were compared to experimental data gathered from the literature. The computational structural and geometric parameters correlate well with the experimental ones derived from the literature. Our UV-visible spectrum results reveal that DPP can work as semiconductors in their own right without being included into other derivative materials, however that halogen derivatives improve mobility and charge transfer. On optimised compound structures, frontier molecular orbital analyses (HOMO and LUMO energies) and global chemical reactivity descriptors were derived.
References
| [1] | S. Qu and H. Tian, “Diketopyrrolopyrrole (DPP)-based materials for organic photovoltaics,” Chem. Commun., vol. 48, no. 25, pp. 3039–3051, 2012, doi: 10.1039/c2cc17886a. |
| [2] | N. O. Tapabashi, K. M. Al-janaby, and S. A. Mohammed, “Thermal Performance, Photostability and UV-Visible Spectroscopic Studies of Some Synthesized azo- Schiff and Bis-azo-Schiff bases,” no. 7, pp. 210–212, 2017. |
| [3] | A. M. Ghaleb and A. Q. Ahmed, “Structural, electronic, and optical properties of sphalerite ZnS compounds calculated using density functional theory (DFT),” Chalcogenide Lett., vol. 19, no. 5, pp. 309–318, May 2022, doi: 10.15251/CL.2022.195.309. |
| [4] | N. Mohammed, S. J. Shakkor, S. M. Abdalhadi, and Y. K. Al-Bayati, “Two multifunctional benzoquinone derivatives as small molecule organic semiconductors for bulk heterojunction and perovskite solar cells,” Main Gr. Chem., vol. 21, no. 4, pp. 943–952, Dec. 2022, doi: 10.3233/MGC-210187. |
| [5] | Q. Liu, S. E. Bottle, and P. Sonar, “Developments of Diketopyrrolopyrrole-Dye-Based Organic Semiconductors for a Wide Range of Applications in Electronics,” Adv. Mater., vol. 32, no. 4, pp. 1–46, 2020, doi: 10.1002/adma.201903882. |
| [6] | R. Almughathawi, S. Hou, Q. Wu, Z. Liu, W. Hong, and C. Lambert, “Conformation and Quantum-Interference-Enhanced Thermoelectric Properties of Diphenyl Diketopyrrolopyrrole Derivatives,” ACS Sensors, vol. 6, no. 2, pp. 470–476, 2021, doi: 10.1021/acssensors.0c02043. |
| [7] | Q. Yang, X. Sun, J. Han, and L. Wang, “Beauty in chemistry: A self-organized and dual-phase emissive diketopyrrolopyrrole derivative as high-yield fluorescent material,” Dye. Pigment., vol. 194, no. July, p. 109655, 2021, doi: 10.1016/j.dyepig.2021.109655. |
| [8] | Q. Liu, S. E. Bottle, and P. Sonar, “Developments of Diketopyrrolopyrrole-Dye-Based Organic Semiconductors for a Wide Range of Applications in Electronics,” Adv. Mater., vol. 32, no. 4, 2020, doi: 10.1002/adma.201903882. |
| [9] | Y. Cai et al., “Small-molecule diketopyrrolopyrrole-based therapeutic nanoparticles for photoacoustic imaging-guided photothermal therapy,” Nano Res., vol. 10, no. 3, pp. 794–801, 2017, doi: 10.1007/s12274-016-1332-2. |
| [10] | X. Wang, B. Jiang, C. Du, X. Ren, Z. Duan, and H. Wang, “Fluorinated dithienyl-diketopyrrolopyrrole: A new building block for organic optoelectronic materials,” New J. Chem., vol. 43, no. 41, pp. 16411–16420, 2019, doi: 10.1039/c9nj04060a. |
| [11] | Z. Yi, S. Wang, and Y. Liu, “Design of High-Mobility Diketopyrrolopyrrole-Based π-Conjugated Copolymers for Organic Thin-Film Transistors,” Adv. Mater., vol. 27, no. 24, pp. 3589–3606, 2015, doi: 10.1002/adma.201500401. |
| [12] | J. Xu et al., “Aryl modification of diketopyrrolopyrrole-based quaternary ammonium salts and their applications in copper electrodeposition,” Dye. Pigment., vol. 170, no. April, p. 107559, 2019, doi: 10.1016/j.dyepig.2019.107559. |
| [13] | J. Humphreys et al., “Solid state structure and properties of phenyl diketopyrrolopyrrole derivatives,” CrystEngComm, vol. 23, no. 8, pp. 1796–1814, 2021, doi: 10.1039/d1ce00039j. |
| [14] | W. W. Bao et al., “Diketopyrrolopyrrole (DPP)-Based Materials and Its Applications: A Review,” Front. Chem., vol. 8, no. September, pp. 1–6, 2020, doi: 10.3389/fchem.2020.00679. |
| [15] | M. Raftani, T. Abram, A. Azaid, R. Kacimi, M. N. Bennani, and M. Bouachrine, “Theoretical design of new organic compounds based on diketopyrrolopyrrole and phenyl for organic bulk heterojunction solar cell applications: DFT and TD-DFT study,” Mater. Today Proc., vol. 45, pp. 7334–7343, 2021, doi: 10.1016/j.matpr.2020.12.1228. |
| [16] | S. M. Wagalgave et al., “Aggregation induced emission (AIE) materials based on diketopyrrolopyrrole chromophore for CdS nanowire solar cell applications,” J. Electroanal. Chem., vol. 895, no. May, p. 115451, 2021, doi: 10.1016/j.jelechem.2021.115451. |
| [17] | M. Raftani, T. Abram, A. Azaid, R. Kacimi, M. N. Bennani, and M. Bouachrine, “Theoretical design of new organic compounds based on diketopyrrolopyrrole and phenyl for organic bulk heterojunction solar cell applications: DFT and TD-DFT study,” Mater. Today Proc., vol. 45, no. xxxx, pp. 7334–7343, 2021, doi: 10.1016/j.matpr.2020.12.1228. |
| [18] | B. Barszcz, K. Kędzierski, H. Y. Jeong, and T. D. Kim, “Spectroscopic properties of diketopyrrolopyrrole derivatives with long alkyl chains,” J. Lumin., vol. 185, pp. 219–227, 2017, doi: 10.1016/j.jlumin.2017.01.019. |
| [19] | Y. Patil and R. Misra, “Rational molecular design towards NIR absorption: Efficient diketopyrrolopyrrole derivatives for organic solar cells and photothermal therapy,” J. Mater. Chem. C, vol. 7, no. 42, pp. 13020–13031, 2019, doi: 10.1039/c9tc03640g. |
| [20] | L. Wang, B. Lai, X. Ran, H. Tang, and D. Cao, “Recent Advances of Diketopyrrolopyrrole Derivatives in Cancer Therapy and Imaging Applications,” Molecules, vol. 28, no. 10, 2023, doi: 10.3390/molecules28104097. |
| [21] | A. Chiminazzo et al., “Diketopyrrolopyrrole Bis-Phosphonate Conjugate: A New Fluorescent Probe for In Vitro Bone Imaging,” Chem. - A Eur. J., vol. 25, no. 14, pp. 3617–3626, 2019, doi: 10.1002/chem.201805436. |
| [22] | Y. Jiang et al., “Multibranched triarylamine end-capped triazines with aggregation-induced emission and large two-photon absorption cross-sections,” Chem. Commun., vol. 46, no. 26, pp. 4689–4691, 2010, doi: 10.1039/c0cc00803f. |
| [23] | J. David, M. Weiter, M. Vala, J. Vyňuchal, and J. Kučerík, “Stability and structural aspects of diketopyrrolopyrrole pigment and its N-alkyl derivatives,” Dye. Pigment., vol. 89, no. 2, pp. 137–143, 2011, doi: 10.1016/j.dyepig.2010.10.001. |
| [24] | S. G. Surya, S. S. Nagarkar, S. K. Ghosh, P. Sonar, and V. Ramgopal Rao, “OFET based explosive sensors using diketopyrrolopyrrole and metal organic framework composite active channel material,” Sensors Actuators, B Chem., vol. 223, pp. 114–122, 2016, doi: 10.1016/j.snb.2015.09.076. |
| [25] | M. Vala, J. Vyňuchal, P. Toman, M. Weiter, and S. Luňák, “Novel, soluble diphenyl-diketo-pyrrolopyrroles: Experimental and theoretical study,” Dye. Pigment., vol. 84, no. 2, pp. 176–182, 2010, doi: 10.1016/j.dyepig.2009.07.014. |
| [26] | D. Cao, Q. Liu, W. Zeng, S. Han, J. Peng, and S. Liu, “Diketopyrrolopyrrole-containing polyfluorenes: Facile method to tune emission color and improve electron affinity,” Macromolecules, vol. 39, no. 24, pp. 8347–8355, 2006, doi: 10.1021/ma0615349. |
| [27] | Y. Zhu, A. R. Rabindranath, T. Beyerlein, and B. Tieke, “Highly luminescent 1,4-diketo-3,6-diphenylpyrrolo[3,4-c]pyrrole-(DPP-) based conjugated polymers prepared upon suzuki coupling,” Macromolecules, vol. 40, no. 19, pp. 6981–6989, 2007, doi: 10.1021/ma0710941. |
| [28] | S. Matsumura et al., “Stability and Utility of Pyridyl Disulfide Functionality in RAFT and Conventional Radical Polymerizations,” J. Polym. Sci. Part A Polym. Chem., vol. 46, no. April, pp. 7207–7224, 2008, doi: 10.1002/pola. |
| [29] | S. Luňák, J. Vyňuchal, and R. Hrdina, “Geometry and absorption of diketo-pyrrolo-pyrrole isomers and their π-isoelectronic furo-furanone analogues,” J. Mol. Struct., vol. 919, no. 1–3, pp. 239–245, 2009, doi: 10.1016/j.molstruc.2008.09.022. |
| [30] | Y. S. Mary et al., “FT-IR, FT-Raman, SERS and computational study of 5-ethylsulphonyl-2-(o- chlorobenzyl)benzoxazole,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 96, pp. 617–625, 2012, doi: 10.1016/j.saa.2012.07.006. |
| [31] | J. Lukose et al., “Synthesis, structural and vibrational investigation on 2-phenyl-N-(pyrazin- 2-yl)acetamide combining XRD diffraction, FT-IR and NMR spectroscopies with DFT calculations,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 135, pp. 608–616, 2015, doi: 10.1016/j.saa.2014.07.004. |
| [32] | Y. S. Mary et al., “Vibrational spectra, NBO analysis, HOMO-LUMO and first hyperpolarizability of 2-{[(2-Methylprop-2-en-1-yl)oxy]methyl}-6-phenyl-2,3,4,5-tetrahydro-1,2,4- triazine-3,5-dione, a potential chemotherapeutic agent based on density functional theory calculations,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 133, pp. 449–456, 2014, doi: 10.1016/j.saa.2014.06.036. |
| [33] | M. Karabacak, M. Çinar, A. Çoruh, and M. Kurt, “Theoretical investigation on the molecular structure, Infrared, Raman and NMR spectra of para-halogen benzenesulfonamides, 4-X-C6H4SO2NH2 (X = Cl, Br or F),” J. Mol. Struct., vol. 919, no. 1–3, pp. 26–33, 2009, doi: 10.1016/j.molstruc.2008.08.007. |
| [34] | V. Arjunan and S. Mohan, “Fourier transform infrared and FT-Raman spectral analysis and ab initio calculations for 4-chloro-2-methylaniline and 4-chloro-3-methylaniline,” J. Mol. Struct., vol. 892, no. 1–3, pp. 289–299, 2008, doi: 10.1016/j.molstruc.2008.05.053. |
| [35] | P. K. Murthy et al., “Synthesis, conformational, characterization and reactivity study of 1,7-bis(4-bromophenyl)heptane-1,7-dione,” J. Mol. Struct., vol. 1175, pp. 269–279, 2019, doi: 10.1016/j.molstruc.2018.08.003. |
| [36] | B. T. Gowda, K. Jyothi, J. Kožíšek, and H. Fuess, “Crystal Structure Studies on p-Substitutedbenzenesulphonamides 4-X-C 6H4SO2NH2 (X = CH3, NH2 F, Cl or Br),” Zeitschrift fur Naturforsch. - Sect. A J. Phys. Sci., vol. 58, no. 11, pp. 656–660, 2003, doi: 10.1515/zna-2003-1110. |
| [37] | M. Vala, M. Weiter, J. Vyňuchal, P. Toman, and S. Luňák, “Comparative studies of diphenyl-diketo-pyrrolopyrrole derivatives for electroluminescence applications,” J. Fluoresc., vol. 18, no. 6, pp. 1181–1186, 2008, doi: 10.1007/s10895-008-0370-x. |
| [38] | E. Normaya, M. N. Ahmad, Y. Farina, and K. H. K. Bulat, “Synthesis, characterization and preliminary study on acetylpyrazine N(4)butylthiosemicarbazone as a potential CDK2 inhibitor combined with DFT calculations,” J. Braz. Chem. Soc., vol. 29, no. 10, pp. 2197–2206, 2018, doi: 10.21577/0103-5053.20180097. |
| [39] | A. Karmakar, P. Bandyopadhyay, S. Banerjee, N. C. Mandal, and B. Singh, “Synthesis, spectroscopic, theoretical and antimicrobial studies on molecular charge-transfer complex of 4-(2-thiazolylazo)resorcinol (TAR) with 3, 5-dinitrosalicylic acid, picric acid, and chloranilic acid,” J. Mol. Liq., vol. 299, p. 112217, 2020, doi: 10.1016/j.molliq.2019.112217. |
| [40] | B. Kosar and C. Albayrak, “Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 78, no. 1, pp. 160–167, 2011, doi: 10.1016/j.saa.2010.09.016. |
| [41] | M. Liu, J. Chen, Y. Chen, and Y. Zhu, “Interaction between smithsonite and carboxyl collectors with different molecular structure in the presence of water: A theoretical and experimental study,” Appl. Surf. Sci., vol. 510, no. January, p. 145410, 2020, doi: 10.1016/j.apsusc.2020.145410. |
| [42] | M. A. Mohammad Alwi et al., “Two-Dimensional Infrared Correlation Spectroscopy, Conductor-like Screening Model for Real Solvents, and Density Functional Theory Study on the Adsorption Mechanism of Polyvinylpolypyrrolidone for Effective Phenol Removal in an Aqueous Medium,” ACS Omega, vol. 6, no. 39, pp. 25179–25192, 2021, doi: 10.1021/acsomega.1c02699. |
| [43] | M. Vala and J. Kraj, “Author ’ s personal copy Dyes and Pigments HOMO and LUMO energy levels of N, N 0 -dinitrophenyl-substituted polar diketopyrrolopyrroles (DPPs)”. |
| [44] | C. B. Nielsen, M. Turbiez, and I. McCulloch, “Recent advances in the development of semiconducting DPP-containing polymers for transistor applications,” Adv. Mater., vol. 25, no. 13, pp. 1859–1880, 2013, doi: 10.1002/adma.201201795. |
| [45] | N. N. Nyangiwe and C. N. M. Ouma, “Adsorption and coadsorption of single and multiple natural organic matter on Ag (1 1 1) surface: A DFT-D study,” Appl. Surf. Sci., vol. 505, no. 111, p. 144609, 2020, doi: 10.1016/j.apsusc.2019.144609. |
| [46] | R. T. Ulahannan et al., “Molecular structure, FT-IR, FT-Raman, NBO, HOMO and LUMO, MEP, NLO and molecular docking study of 2-[(E)-2-(2-bromophenyl)ethenyl]quinoline-6-carboxylic acid,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 151, pp. 184–197, 2015, doi: 10.1016/j.saa.2015.06.077. |