-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Condensed Matter Physics
p-ISSN: 2163-1115 e-ISSN: 2163-1123
2024; 13(2): 21-34
doi:10.5923/j.ajcmp.20241302.01
Received: Jun. 26, 2024; Accepted: Jul. 24, 2024; Published: Jul. 27, 2024

Computational Study of Electron-Withdrawing and Donating Groups Effects on Structure and Electronic Properties of the 2-(2-Methylphenyl)-,4-,5-Diphenyl- 1H-Imidazole Compound
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLHindreen A. Ibrahim
Basic Sciences Branch, College of Dentistry, University of Kirkuk, Iraq
Correspondence to: Hindreen A. Ibrahim, Basic Sciences Branch, College of Dentistry, University of Kirkuk, Iraq.
| Email: |  |
Copyright © 2024 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

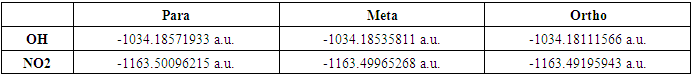
In the current study, the 2-(2-methylphenyl)-,4-,5-diphenyl-1H-imidazole based (donor (OH) and accepter (NO2) groups) are examined in order to determine the impact of various substituents on the optical and structural properties using density functional theory (DFT) with a global hybrid functional (B3LYP) and 6-311G(d,p) as a basis set. The geometry optimization of 2-(methylphenyl)-,4-,5-diphenyl-1H-imidazole with (donor (OH) and accepter (NO2) groups substitutes in ortho, meta, and para positions have been calculated in the gas phase. It was observed that the two substituted groups exhibited a minimal potential energy distribution (PED) for molecules in the para position. It is observed that the incorporation of an electron withdrawing group (NO2) at the para position and an electron donating group (OH) at the para positions of 2-(2-methylphenyl)-,4-,5-diphenyl-1H-imidazole system resulted in a greater shift towards the blue end of the absorption spectrum. The TD-DFT technique was used to forecast the UV-Vis spectral parameters, including the maximum wavelength, energy, and oscillator strength, of three compounds. The molecule containing a para-positioned NO2 group demonstrated the longest absorption wavelength at λ= 395.13 nm. The HOMO-LUMO energy gaps, global reactivity descriptors and Molecular electrostatic potential surfaces (MEPS) are computed to examine how various substituents impact the structural and optical characteristics of 2-(2-methylphenyl)-,4-,5-diphenyl-1H-imidazole.
Keywords: Imidazole, Withdrawing and donating groups, DFT, Electronic structure, HOMO-LUMO Energy gap, Global reactivity descriptors
Cite this paper: Hindreen A. Ibrahim, Computational Study of Electron-Withdrawing and Donating Groups Effects on Structure and Electronic Properties of the 2-(2-Methylphenyl)-,4-,5-Diphenyl- 1H-Imidazole Compound, American Journal of Condensed Matter Physics, Vol. 13 No. 2, 2024, pp. 21-34. doi: 10.5923/j.ajcmp.20241302.01.
Article Outline
1. Introduction
- Recent years have seen remarkable progress in organic semiconductors, which provide several benefits such reduced weight, scalability, simplicity of synthesis, affordability, and adaptability [1,2], as well as recently, nonlinear optical (NLO) materials have become increasingly significant in fields such as optical data storage, communication systems, high-resolution spectroscopy, laser printing, optical signal processing, remote sensing, and optical signal processing [3,4]. Polar organic compounds have a broad variety of physical and chemical characteristics, making their synthesis and assessment crucial. In most cases, the 𝜋-electron conjugated structure is what makes up organic compounds. Under the influence of an external electric field, the π-electron moiety's conjugation creates a route that continues over the whole length of the conjugation process. Improving the asymmetric electronic distribution in both the ground and excited states can lead to an enhancement in the NLO characteristics, which can be achieved by fictionalizing the π bond systems with appropriate electron donor and acceptor groups at both ends [5]. The NLOphoric molecules structure, which have donor and acceptor groups connected by a conjugated bridge (D-π-A) system [6]. Nonlinear optics (NLO) is currently a focal point of scientific investigation due to its critical role in enabling emerging technologies in fields including signal processing, optical interconnections, and telecommunications to perform the following operations: frequency shifting, optical modulation, optical switching, optical logic, and optical memory [7]. These materials have garnered significant attention due to their remarkable NLO properties, which include a rapid response in the electro-optic effect and large second or third order hyperpolarizabilities, surpassing those of inorganic NLO materials [8]. Heterocyclic compounds are significant contenders with diverse structural attributes for a wide range of biological, pharmaceutically active substances, and agrochemicals [9]. Imidazole is a significant group of heterocyclic compounds due to their remarkable biological activity and their application in synthetic, medicinal, and pharmaceutical chemistry. Imidazole is a heterocyclic molecule with a five-membered aromatic structure and two nitrogen atoms located at the 1, 3- position. The structure in question is referred to as 1, 3-diazole in a systematic manner. It is a strongly polar chemical that exhibits a high degree of solubility in water. It has amphoteric properties, meaning it can act as both an acid and a base. Its basicity is 100 times more than that of pyridine [10], which can be discovered in various natural substances [11]. Thus, the development of easy artificial methods for these nitrogen-containing heterocyclic molecules using readily available reagents is a critical undertaking in the field of organic synthesis [12]. In this context, multicomponent reactions (MCRs) occupy a highly advantageous position, allowing the creation of diverse bonds by simultaneously combining three or more reactants in a single step [13]. The investigation of fused polycyclic aromatic molecules has garnered attention due to their numerous industrial and scientific applications, in addition to their fundamental electronic structure. Polycyclic aromatic compounds are utilized as "host and dopant materials" in organic light-emitting diodes (OLEDs), molecular probes, fluorescent labels, and light-emitters. One of the critical factors in adjusting the electronic properties of polycyclic aromatic rings is the inclusion of an electronegative nitrogen atom within them. The -N atom's elevated electronegativity contributes to the reduction of the "LUMO" level in N-heteroaromatic compounds, facilitating electron injection and enhancing charge carrier stability [6]. Multicomponent reactions offer numerous benefits, including reduced time intervals, cheaper costs, less energy usage, high yields, quicker reaction times, and simplified purification methods, while avoiding the need for intermediate separation [11]. Photo induced intramolecular proton transfer events can be observed in aromatic and heteroaromatic compounds with strong O-H and N-H intramolecular hydrogen bonds [14]. Imidazole’s are widely prevalent ring systems in heterocyclic chemistry, extensively studied in scientific literature for their synthesis, characteristics, and diverse uses across several disciplines. Imidazole’s are frequently found in marine natural products and present notable difficulties in their synthesis. They also play a crucial role in the biological activity observed in these items. The imidazole ring is well recognized as a significant structure in medicinal chemistry and is present in numerous therapeutic medicines, including angiotensin II inhibitors [15]. The utilization of modeling approaches, specifically molecular docking, has greatly enhanced drug discovery by promoting the development of imidazole scaffolds as a common pharmacophore. Prior to designing a new molecule, chemists utilize molecular modeling and computational studies to concentrate on synthesizing moieties of pharmacological significance. Furthermore, comprehensive computer analyses using manufactured molecules enable chemists to produce novel pharmacologically significant compounds of a similar nature [16]. Imidazole and its derivatives have extensive use as intermediates in the synthesis of many organic target molecules, such as medicines, photographic chemicals, agrochemicals, dyes, epoxy curing agents, corrosion inhibitors, plastic modifiers, and adhesives [17]. The imidazole nucleus is the central structure found in several important components of the human body, such as the amino acid vitamin B12, histidine, a building block of DNA bases, biotin, purines, and histamine. It is also found in the composition of numerous natural or synthesized medicinal compounds, including as cimetidine, metronidazole, and azomycin [18]. Imidazole derivatives offer significant benefits for synthesizing many types of pharmaceutical molecules with excellent therapeutic efficacy. They exhibit analgesic [12], anti-mycotic, anti-allergy [19], anti-bacterial [20], anti-inflammatory [21], anti-viral, therapeutic activity [22], anti-ulcerative, and anti-biotic [23], antitumor [24], anti-microbial [25], anticonvulsant [26], cytotoxic [27], anticancer [28], fine chemicals [29], anthelmintic, anti-parasitic, antiepileptic agents, antiplatelet drugs, and activity inhibition of glucagon receptor [12]. Imidazole also shows strong efficacy against RNA viruses, human cytomegalovirus (HCMV), HIV and herpes (HSV-1) [30], exhibit potent α-glucosidase inhibitory activity [31] Heterocyclic imidazole derivatives have garnered significant interest due to their distinctive optical characteristics and their utility in synthesizing functionalized materials [18]. Numerous 2,4,5-trisubstituted imidazole’s are fungicides [23], herbicides [29] and regulators of plant growth. Additionally, they function as p38 MAP kinase inhibitors [19]. They also have a vital function in the field of material science due to their widespread application as materials that emit blue light, agents used for fluorescence labeling, and chemical sensors. Various synthetic methods have been devised for the production of substituted imidazole’s, owing to their wide range of uses [32]. Conversely, imidazole derivatives have demonstrated their efficacy as very effective fluorophores owing to their extensive π-conjugated structure and the acidic properties of the NH- group. The utilization of these optical characteristics has been implemented in a variety applications, including, luminescent materials, two-photon two-photon fluorescence microscopy, absorption applications, photographic materials, 3D microfabrication and high density storage, Non Linear Optics (NLO), Organic Light Emitting Diodes (OLEDs), Dye Sensitized Solar Cells (DSSC) and molecular switches [33]. The NLO materials have garnered significant attention due to their applicability in optoelectronic materials and optical switching. The optical properties of organic nonlinear optical (NLO) material arise only from electrical factors [6]. Furthermore, the molecular structure's nature plays a crucial role in the creation of every crystal system. In order to obtain the necessary guidance from established crystal structures, the crystal engineer frequently relies on crystal packing diagrams and the precise measurement of bond lengths and angles [34]. Here, theoretical methods provide useful insights into molecular conformation, electronic structure, electronic , and optical characteristics [6].The current study, employs molecular electronic structure calculations, specifically density functional theory (DFT) and Time-dependent density functional theory (TD-DFT), to investigate the impact of different functional groups on the electronic structure and optical properties of 2-(methylphenyl)-,4-,5-diphenyl-1H-imidazole and its derivatives. The detailed structural optimization of 2-(2-methylphenyl)-,4-,5-diphenyl-1H-imidazole with donor (OH) and accepter (NO2) groups substitutes in ortho, meta and para positions have been calculated by Density Functional Theory method DFT/B3LYP with 6-311G(d,p) as a basis set in the gas phase. For the optimal geometry in vacuum, the vibrational frequency was determined in order to find the global minima on the potential energy surface. It was observed that the two substituted groups exhibited a minimal potential energy distribution (PED) for molecules in the para position as shown in Table 1. As a result, the acceptor (NO2) and donor (OH) group substitutes in para positions were selected for all calculations utilizing the identical method and basis set that were utilized to optimize the molecular geometry. Theoretical structural parameters were compared to experimental values obtained from literature. The UV-Vis spectrum, energy, and oscillator strength were determined using TD-DFT computations. The compounds have been analyzed using HOMO-LUMO analysis and Molecular electrostatic potential surfaces (MEPS) to illustrate the transfer of charge.
|
2. Computational Details
- In this study, the compounds were analyzed using quantum chemical methods based on density functional theory (DFT). Specifically, Becke's three parameters (B3LYP) hybrid exchange correlation with Lee-Yang-Parr functional was performed, along with a 6-311G basis set. The properties of all types were calculated in the gas phase for this research. The computer programs Gaussian09 and Gauss-View, which allow for molecular visualization, are utilized for all quantum chemical computations [35]. By solving the self-consistent field equation effectively, the equilibrium geometry which represents the lowest energy state on the Potential Energy Surface (PES) was achieved [36] to calculate the bond lengths, bond angles and dihedral angles [35] as well as perform Frontier molecular orbital computations (FMOs). For optimal optimized geometry in vacuum, the vibrational frequency was determined the potential energy surface's global minima [34]. In addition, the TD-DFT approach was used to calculate UV-Vis spectra, electronic transitions, oscillator strengths and vertical excitation energies. The time-dependent DFT method was used to ascertain the electronic characteristics, including the HOMO and LUMO energies [34]. In order to investigate the molecular stability of the compounds, we utilized density functional theory DFT. We have successfully obtained significant data regarding the bond properties, molecular orbitals, and absorption pattern of the product molecule. Initially, a conceptual framework was devised, using the conventional chemical intuition, utilizing a computational tool. Subsequently, a theoretical analysis was conducted on the simulated structure in order to provide molecular data [32]. To determine Included in Gaussian is Bernhard Schlegel's "Berny algorithm" for optimization. Using the atomic forces and the second derivative matrix, this program optimizes the molecular structure to find the next local minimum on the potential energy surface by predicting energetically better structures. Because it is expensive to explicitly calculate the second derivative matrix, the Berny algorithm uses a simple valence force field to build an approximate Hessian at the start of the optimization process. As the optimization progresses, it updates this approximate Hessian with the energies and first derivatives calculated [37].
3. Result and Discussion
3.1. Molecular Geometries
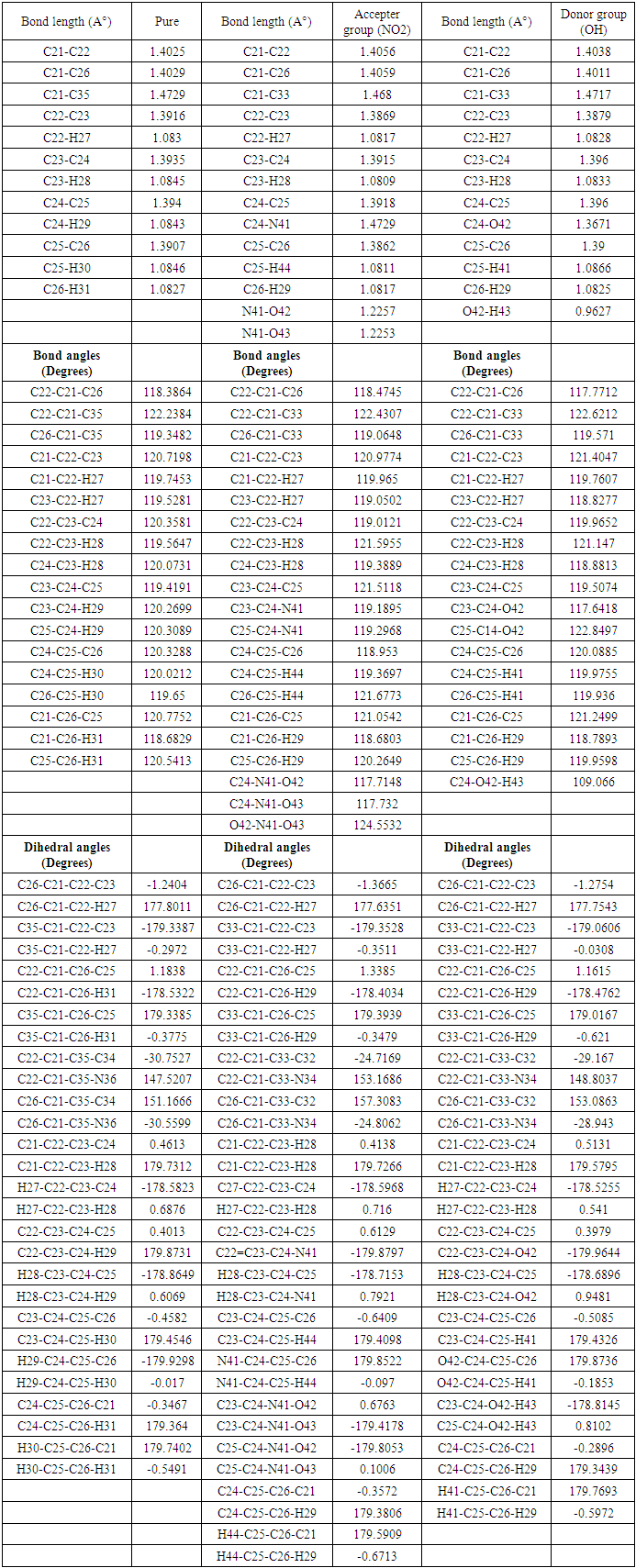
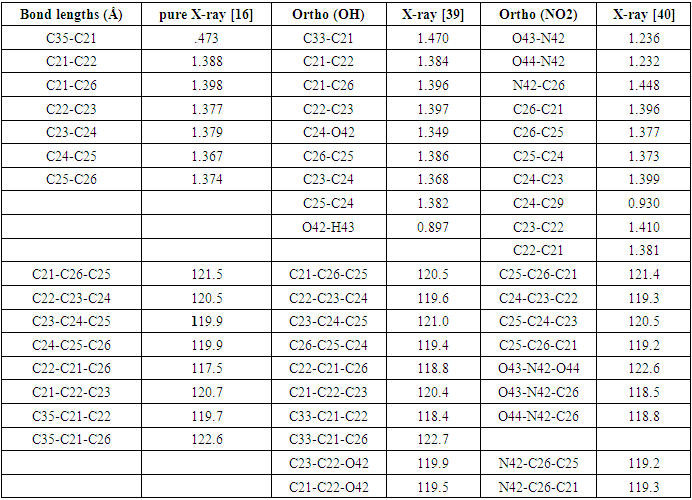
- The geometry optimization process is a critical stage in the determination of standard electrode potentials [38]. The optimal structure is determined using the Density Functional Theory (DFT) with Gaussian09 computer package, which employs the B3LYP (Becke three parameter Lee-Yang-Parr) approach with a 6-311G basis set [35]. The parameters of the structure have been optimized, the bond lengths, bond angles and dihedral angles in the para position, additionally the X-ray data obtained experimentally [15,38,39] are listed in Table 2 and 3 respectively, in accordance with the atom numbering scheme given in Fig. 1. Table 2 and 3 reveals that the calculated bond lengths are all slightly greater than the experimental values, as are most of the bond angles. The inconsistencies can be attributed to the assumption that the computations are based on an isolated molecule, neglecting the intermolecular Columbic contact with nearby molecules. It is widely recognized that DFT approaches consistently overestimate bond lengths, especially those involving carbon and hydrogen (C–H) bonds [41]. The overestimation is corroborated in our estimate, as evidenced by Table 2. Theoretical C–H bond lengths exceed 1.0 Å, however the experimental results indicate lesser values [15,38,39,41].
|
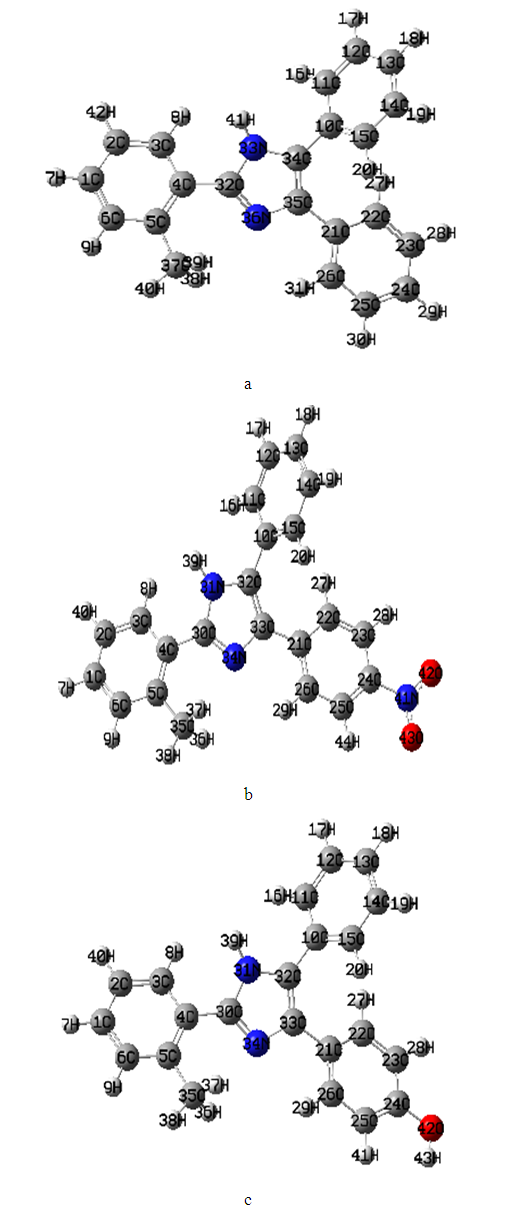 | Figure 1. The theoretical molecular geometry of the (a) 2-(2-methylphenyl), 4,5-diphenyl-1H-imidazole, (b) 2-(2-methylphenyl), 4-(4-nitrophenol), phenyl-1H-imidazole, (c) 2-(2-methylphenyl), 4-(4-hydroxyphenol), phenyl-1H-imidazole with atom numbering where (gray, white, blue and red is the carbon C, hydrogen H, nitrogen N and oxygen O) respectively |
3.2. Electronic Properties and UV/Vis Absorption
- The electronic transitions of the compounds under study were calculations by Time Dependent-DFT method [34] using the Becke three parameter Lee-Yang-Parr (B3LYP) exchange correlation functional with 6-311G(d,p) as a basis set in the gas phase. The Gaussain09W computer package was employed for these calculations, and no restrictions were placed on the geometry [5]. Whether crystalline substances exhibit direct or indirect optical transitions is determined by their band structure. In the case of a direct transition, n is either 1/2 or 3/2, contingent on whether the transition is permitted or prohibited in the context of quantum mechanics. In a similar fashion, n=2 or 3 denotes an indirect form of permitted and prohibited transition [5]. The UV–Vis–NIR spectral analysis is crucial for determining the optical characteristics of materials and measuring their absorbance within the suitable wavelength range. The occurrence of this spectrum is a result of the molecule's electronic transitions (σ, n, π) from the lowest energy state to higher energy levels. Figure 2 displays the absorption spectra in the ultraviolet-visible-near infrared (UV-Vis-NIR) range. The absorbance spectrum revealed distinct absorbance properties in the ultraviolet (UV) and visible regions, spanning around 250 to 450 nm. Beyond this range, no absorbance was seen in the near-infrared (NIR) area. The absorbance observed in the crystal may be attributed to the electronic transition occurring from a 'non-bonding' (n) orbital to a 'antibonding' (π*) orbital, which is represented as (n→π*) [43]. The absorption wavelength (λ), excitation energies E and oscillator strength (f), are shown in Table 4. Calculation of theoretical absorption spectra was performed at the lowest energy points of the ground state. The determined energy values for the bandgap (Egap) of the absorption band a range of 300 nm to 329 nm, 310 nm to 340 nm while the accepter compound has two bands in a range 310 nm to 330 nm and 375 nm to 405 nm. The Egap values were observed around ∼3.00 eV for different molecules. According to the computed absorption spectra, the electronic transition from the HOMO to the LUMO occurs at the highest absorption wavelength with 98% for 2-(2-methylphenyl), 4,5-diphenyl-1H-imidazole contribution. Table 4 shows the other wavelengths and their corresponding estimated values that have significant contributions. TD-DFT calculations anticipate the presence of three transitions inside the UV-Vis range for three distinct molecules. The primary correlation of the strong electronic transitions occurs between the ground state and the first excited state is mainly related the HOMO (MO:82) →LUMO (MO:83) excitation at 3.7651eV (329.30nm) with an oscillator strength f=0.3962a.u and HOMO (MO:86) →LUMO (MO:87) excitation at 3.6184eV (342.65nm) with an oscillator strength f=0.3147a.u in gas phase are assigned to π→π* transition for 2-(2-methylphenyl), 4,5-diphenyl-1H-imidazole, 2-(2-methylphenyl), 4-(4-hydrophenyl), 5-phenyl-1H-imidazole, except the 2-(2-methylphenyl), 4-(4-nitrophenyl), 5-phenyl-1H-imidazole the strong electronic transitions occurred from the ground state to the second excited state is mainly related the HOMO (MO:93) →LUMO+1 (MO:95) excitation at 3.9496eV (313.92nm) with an oscillator strength f=0.4598a.u, besides, HOMO (MO:93) →LUMO (MO:94) excitation has also contributed though at 3.1378eV (395.13nm) with an oscillator strength f=0.2733a.u it is not a big contribution in gas phase are assigned to π→σ and π→π* transition respectively . For a direct transition n = 1/2 or 3/2 depending on whether the transition is allowed or forbidden in quantum mechanical sense. Similarly, n = 2 or 3 for indirect allowed and forbidden transitions respectively [8]. The π–π* transitions are expected to occur relatively at lower wavelength, due to the consequence of the extended aromaticity of the benzene ring. The computed UV method predicts three electronic transition at 329.30, 301.91, 294.10 with oscillator strength f = 0.3962, 0.2913 and 0.0611a.u., 342.65, 308.84, 303.84nm with oscillator strength f = 0.3147, 0.1843 and 0.1362a.u. and 395.13, 326.36, 313.92 nm with oscillator strength f = 0.2733, 0.0001 and 0.4598a.u. in gas phase for 2-(2-methylphenyl), 4,5-diphenyl-1H-imidazole, 2-(2-methylphenyl), 4-(4-hydrophenyl), 5-phenyl-1H-imidazole and 2-(2-methylphenyl), 4-(4-nitrophenyl), 5-phenyl-1H-imidazole respectively. Table 4 presents the absorption maxima, denoted as ϻmax, which have been computed in relation to the electron availability. The absorption maxima of these molecules are determined by calculations of molecular orbital geometry to be electron transitions between frontier orbitals, specifically translation from HOMO to LUMO. As show from the UV–vis spectra absorption maxima values observed that are 342.65, 329.30 and 313.92 nm for 2-(2-methylphenyl), 4,5-diphenyl-1H-imidazole, 2-(2-methylphenyl), 4-(4-hydrophenyl), 5-phenyl-1H-imidazole and 2-(2-methylphenyl), 4-(4-nitrophenyl), 5-phenyl-1H-imidazole respectively. Since the λmax is influenced by substitution, an increase in the donor character of the substitution results in a greater number of electrons being forced into the molecule, thereby increasing the λmax. High delocalization of π-electrons may account for the visible bands detected at 342.65, 329.30, and 313.92 nm for the aforementioned compounds. The presence of these bands could potentially be attributed to the electronic transition of π→π*. Figure 2 illustrates the UV–vis spectra that was calculated [44].
|
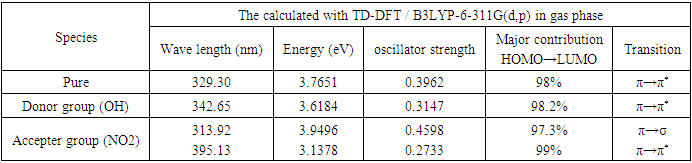
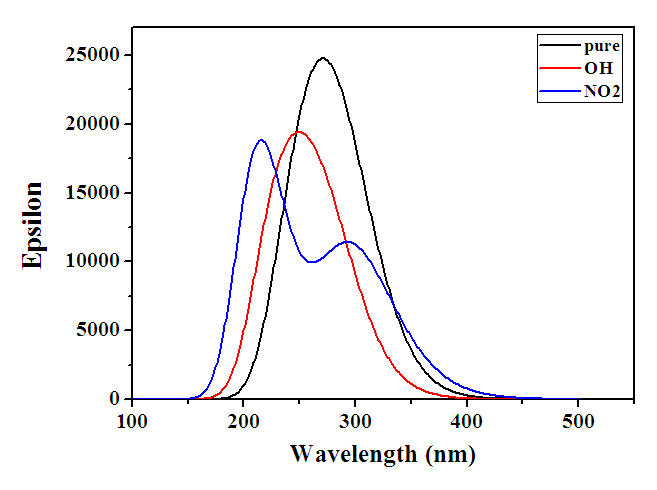 | Figure 2. UV-VIS absorption spectra of 2-(2-methylphenyl), 4,5-diphenyl-1H-imidazole and its derivatives |
3.3. Frontier Molecular Orbital Analysis
- The current study predicts the HOMO and LUMO energies using the B3LYP technique and the 6-31G(d,p) basis sets. Accordingly, the molecules contains 82, 86 and 93 occupied molecular orbitals and 458, 468 and 495 unoccupied virtual molecular orbitals for 2-(2-methylphenyl), 4,5-diphenyl-1H-imidazole and 2-(2-methylphenyl), 4-(4-hydroxyphenyl), 5-phenyl-1H-imidazole and 2-(2-methylphenyl), 4-(4-nitrophenyl), 5-phenyl-1H-imidazole respectively [45]. The highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO) are two significant frontier orbitals that have a vital impact on determining many properties of a compound and are fundamental in the field of quantum chemistry [16]. Researchers in the fields of physics and chemistry find the HOMOs and LUMOs to be of great use. This is due to the fact that the energy gap between these orbitals represents the minimum energy needed to promote an electron, making it one of the most common and consequential energy transfer mechanisms in a system [46]. Chemical reactivity, kinetic stability, optical and electrical properties are all measured by the energy difference between HOMO and LUMO [16]. Furthermore, lower energy gap between the (HOMO) and (LUMO) accounts for the definitive charge transfer interactions occurring within the molecule [39]. When presented in a qualitative graphical format, molecular orbitals can offer valuable insights into the characteristics of reactivity as well as certain structural and physical attributes of molecules [36]. The amplitudes of the (HOMO) and (LUMO) are utilized to identify the significant regions involved in electronic transitions inside molecular orbitals. Additionally, they can be employed to forecast the chemical reactivity of molecular systems [47]. The spectroscopic characteristics of p-conjugated molecular systems and the potential locations for intra- or intermolecular interactions (or reactions) are revealed by the shapes of frontier molecular orbitals. This is because molecular orbital theory provides an explanation for the interactions between the HOMO and LUMO, such as UV-Vis spectral properties [48]. The observed transition from HOMO to LUMO is π→π* [39]. Koopmans's theorem in molecular orbital theory states that the energy of the (EHOMO) is directly connected to the ionization potential (IP) by
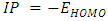 [40], the energy required to liberate the valence electron from an unbound gaseous atom and transform it into a cation can be calculated from its border molecular orbitals; the LUMO energy (ELUMO) has been utilized to gauge the electron affinity by subtracting
[40], the energy required to liberate the valence electron from an unbound gaseous atom and transform it into a cation can be calculated from its border molecular orbitals; the LUMO energy (ELUMO) has been utilized to gauge the electron affinity by subtracting 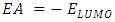 [40], it is the quantity of energy that occurs when an electron is added or consumed to a neutral atom or molecule in the gaseous state to generate a negative ion. It may be determined using the (HOMO) and (LUMO) [16]. Orbitals also give useful information on electron densities, which can be used to find out which molecular region is involved in an energy transfer even the most [46]. Figure 3 shows the three-dimensional plots of significant molecular orbitals, which were used to better understand the bonding schemes of the three compounds contour surfaces. By classifying green as negative and red as positive, these surfaces indicate that the HOMO orbital is highly localized over the all molecule rings. No change occurred in HOMO shade when the donating group (OH) was substituted, whereas the HOMO is not localized over the 4-(4-nitrophenyl) ring and (C22) when the withdrawing group (NO2) was substituted [40]. Therefore, the presence of a highly delocalized (HOMO) indicates that the electrons within the molecule can travel more easily, leading to enhanced intramolecular charge transfer [49] On the other hand, LUMO strongly localized over molecule rings except for a benzene ring covering it lightly, after (OH) substituted LUMO orbital disappeared completely over the hydroxyphenyl ring, while as (NO2) substituted LUMO localized strongly only over the 4-(4-nitrophenyl) ring. According to prior researches by many authors, the capacity of heteroatoms to adsorb onto material surfaces through donor-acceptor interaction is directly proportional to their negative charge. Inhibitors' varying degrees of efficacy are due to the existence of electronegative atoms such as N and O [50]. Typically, an atom that has a higher density of the (HOMO) would have a greater ability to lose an electron. Conversely, an atom with a higher occupation of the (LUMO) will have a greater ability to receive an electron [45], due to electron withdrawing effect of NO2 group majority of electron density is accumulated on N (42) and O (43)/O (44) atoms of NO2 group [50]. Signaling the existence of advantageous atomic sites inside the molecules for potential nucleophilic assaults and its bioactivity that enhances the material's non-linear behavior and molecular reactivity [51]. A small HOMO–LUMO gap distinguishes conjugated molecules due to a substantial level of intramolecular charge transfer from the electron-donor groups at the ends to the electron-acceptor groups along the p-conjugated pathway [36]. If an inhibitor has a high EHOMO value, it will be simpler for it to donate electrons to empty molecular d-orbitals, which means that the inhibitor's ability to donate electrons to the molecular surface is proportional to this value. Meanwhile, ELUMO levels (i.e., a lower value) are linked to the electron-accepting capabilities of inhibitor compounds. It will be easier for inhibitor molecules to accommodate the extra negative charge that is provided by the filled d-orbital. The energy differential (ΔE) between the HOMO and LUMO states is also connected to the stability index of any inhibitor. Reducing the ΔE values will increase the stability of the contact between the inhibitor and the surface [50]. From Table 5, it is evident that EHOMO value of 2-(2-methylphenyl), 4,5-diphenyl-1H-imidazole is -5.46eV it decreased after (OH) group substituted -5.27eV while it increased as (NO2) substituted to -5.91 eV, which indicates the largest inhibitory efficiency is provided by the donating group (OH), which has the strongest electron donation ability and will adsorb to a greater extent. In the same way ELUMO value of original compound is -1.27eV after (OH) substituted it increased to -1.23eV whereas with (NO2) substituted ELUMO value decreased to -2.41eV, lower value of withdrawing group (NO2) indicates its ability to accept electrons [52]. Inspection of the data in Table 5 revealed that ΔE of original compound is 4.19eV after donating group (OH) and withdrawing group (NO2) substituted it become 4.04 eV and 3.49 eV respectively, it shows the withdrawing group (NO2) is lowest, which indicate that they possess a greater capacity for adsorption than the donating group (OH). Two descriptors—hardness and softness—are utilized to quantify the chemical reactivity of a molecule. These descriptors are derived from the HOMO and LUMO energy values using Equations (3) and 4 [16]. The frontier molecular orbital energies yield various global descriptors, including the electrophilic index, maximum charge transfer capability, global hardness, electronegativity, and electronic chemical potential. These descriptors, listed in Table 5, offer valuable insights into the physical and physicochemical properties of the molecule. Currently, a molecule with a lower energy gap and higher global hardness is considered to be more reactive. The electrophilic index, which is determined by the electronic chemical potential and global hardness, indicates the molecule's ability to take electrons from its surroundings [48]. The values corresponding to (Eg), (η), (μ), (χ) and (S) were calculated as specified above and found to be 4.19, 2.1, -3.37, 3.37, 0.24 eV for original compound respectively when donating group (OH) and withdrawing group (NO2) substituted it becomes 4.04, 2.02, -3.25, 3.25, 0.25 eV and 3.49, 1.75, -4.16, 4.16, 0.28 eV for donating group (OH) and withdrawing group (NO2) respectively. This mean the (NO2) group increase the molecule's physicochemical and physical characteristics. We used the following formulae to calculate the chemical potential, electronegativity, global hardness, global softness, global electrophilic index, and extra electronic charge [16].
[40], it is the quantity of energy that occurs when an electron is added or consumed to a neutral atom or molecule in the gaseous state to generate a negative ion. It may be determined using the (HOMO) and (LUMO) [16]. Orbitals also give useful information on electron densities, which can be used to find out which molecular region is involved in an energy transfer even the most [46]. Figure 3 shows the three-dimensional plots of significant molecular orbitals, which were used to better understand the bonding schemes of the three compounds contour surfaces. By classifying green as negative and red as positive, these surfaces indicate that the HOMO orbital is highly localized over the all molecule rings. No change occurred in HOMO shade when the donating group (OH) was substituted, whereas the HOMO is not localized over the 4-(4-nitrophenyl) ring and (C22) when the withdrawing group (NO2) was substituted [40]. Therefore, the presence of a highly delocalized (HOMO) indicates that the electrons within the molecule can travel more easily, leading to enhanced intramolecular charge transfer [49] On the other hand, LUMO strongly localized over molecule rings except for a benzene ring covering it lightly, after (OH) substituted LUMO orbital disappeared completely over the hydroxyphenyl ring, while as (NO2) substituted LUMO localized strongly only over the 4-(4-nitrophenyl) ring. According to prior researches by many authors, the capacity of heteroatoms to adsorb onto material surfaces through donor-acceptor interaction is directly proportional to their negative charge. Inhibitors' varying degrees of efficacy are due to the existence of electronegative atoms such as N and O [50]. Typically, an atom that has a higher density of the (HOMO) would have a greater ability to lose an electron. Conversely, an atom with a higher occupation of the (LUMO) will have a greater ability to receive an electron [45], due to electron withdrawing effect of NO2 group majority of electron density is accumulated on N (42) and O (43)/O (44) atoms of NO2 group [50]. Signaling the existence of advantageous atomic sites inside the molecules for potential nucleophilic assaults and its bioactivity that enhances the material's non-linear behavior and molecular reactivity [51]. A small HOMO–LUMO gap distinguishes conjugated molecules due to a substantial level of intramolecular charge transfer from the electron-donor groups at the ends to the electron-acceptor groups along the p-conjugated pathway [36]. If an inhibitor has a high EHOMO value, it will be simpler for it to donate electrons to empty molecular d-orbitals, which means that the inhibitor's ability to donate electrons to the molecular surface is proportional to this value. Meanwhile, ELUMO levels (i.e., a lower value) are linked to the electron-accepting capabilities of inhibitor compounds. It will be easier for inhibitor molecules to accommodate the extra negative charge that is provided by the filled d-orbital. The energy differential (ΔE) between the HOMO and LUMO states is also connected to the stability index of any inhibitor. Reducing the ΔE values will increase the stability of the contact between the inhibitor and the surface [50]. From Table 5, it is evident that EHOMO value of 2-(2-methylphenyl), 4,5-diphenyl-1H-imidazole is -5.46eV it decreased after (OH) group substituted -5.27eV while it increased as (NO2) substituted to -5.91 eV, which indicates the largest inhibitory efficiency is provided by the donating group (OH), which has the strongest electron donation ability and will adsorb to a greater extent. In the same way ELUMO value of original compound is -1.27eV after (OH) substituted it increased to -1.23eV whereas with (NO2) substituted ELUMO value decreased to -2.41eV, lower value of withdrawing group (NO2) indicates its ability to accept electrons [52]. Inspection of the data in Table 5 revealed that ΔE of original compound is 4.19eV after donating group (OH) and withdrawing group (NO2) substituted it become 4.04 eV and 3.49 eV respectively, it shows the withdrawing group (NO2) is lowest, which indicate that they possess a greater capacity for adsorption than the donating group (OH). Two descriptors—hardness and softness—are utilized to quantify the chemical reactivity of a molecule. These descriptors are derived from the HOMO and LUMO energy values using Equations (3) and 4 [16]. The frontier molecular orbital energies yield various global descriptors, including the electrophilic index, maximum charge transfer capability, global hardness, electronegativity, and electronic chemical potential. These descriptors, listed in Table 5, offer valuable insights into the physical and physicochemical properties of the molecule. Currently, a molecule with a lower energy gap and higher global hardness is considered to be more reactive. The electrophilic index, which is determined by the electronic chemical potential and global hardness, indicates the molecule's ability to take electrons from its surroundings [48]. The values corresponding to (Eg), (η), (μ), (χ) and (S) were calculated as specified above and found to be 4.19, 2.1, -3.37, 3.37, 0.24 eV for original compound respectively when donating group (OH) and withdrawing group (NO2) substituted it becomes 4.04, 2.02, -3.25, 3.25, 0.25 eV and 3.49, 1.75, -4.16, 4.16, 0.28 eV for donating group (OH) and withdrawing group (NO2) respectively. This mean the (NO2) group increase the molecule's physicochemical and physical characteristics. We used the following formulae to calculate the chemical potential, electronegativity, global hardness, global softness, global electrophilic index, and extra electronic charge [16].  | (1) |
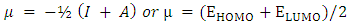 | (2) |
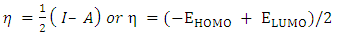 | (3) |
 | (4) |
 | (5) |
 | (6) |
|
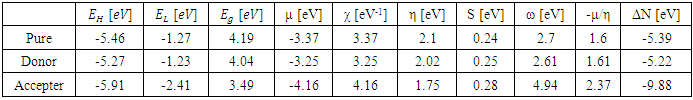
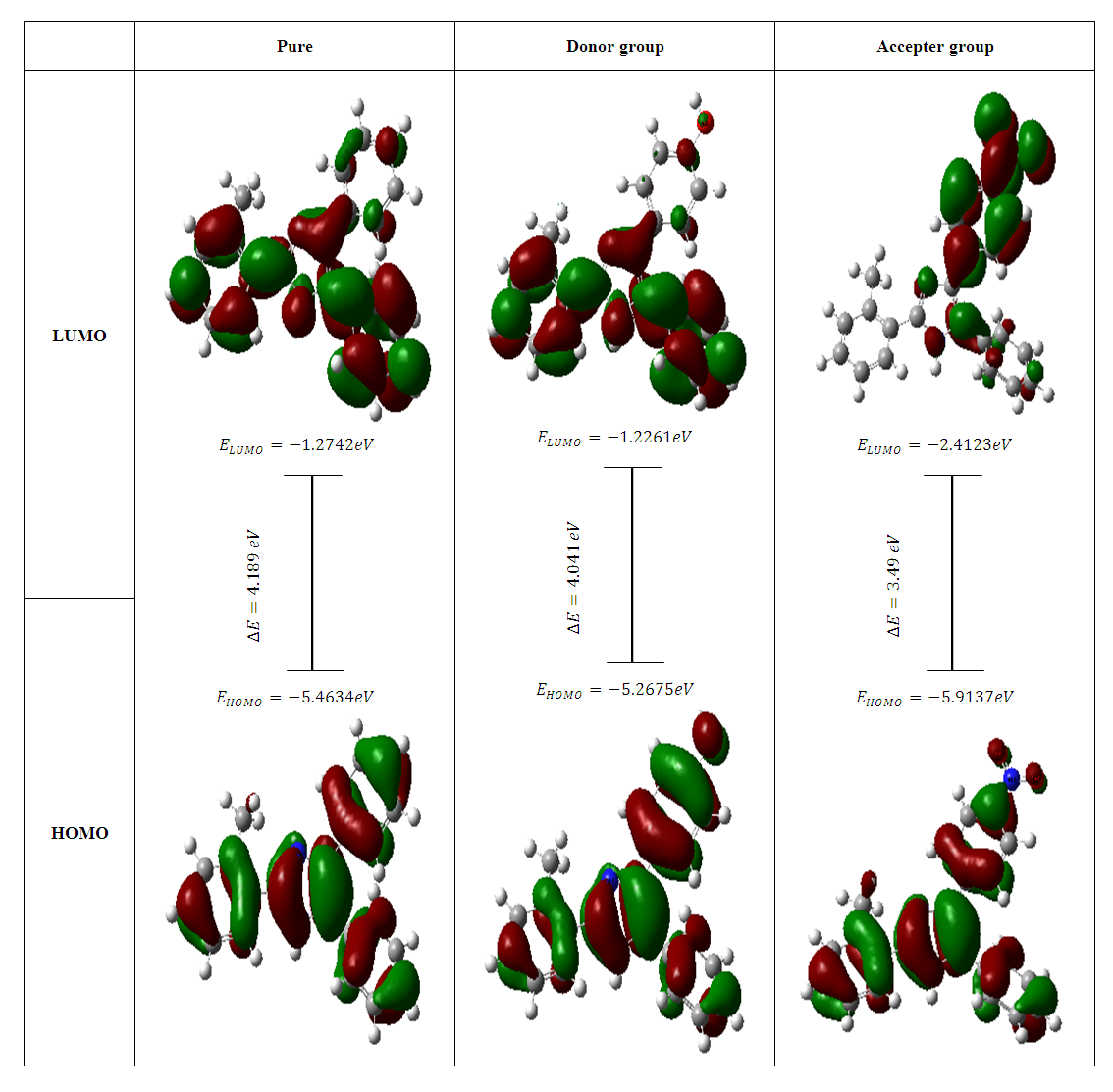 | Figure 3. Molecular orbitals energies |
3.4. Molecular Electrostatic Potential Surfaces (MEPS)
- The electrostatic potential arises from the interactions between the nuclei and electrons within a molecule at various points in its surrounding space, primarily due to electron density. Molecular electrostatic potential serves as a significant tool for assessing and forecasting a molecule's reactivity to electrophilic attacks. It has also been identified as a measure of a molecule's capacity for hydrogen bonding interactions [34]. The Molecular Electron Pair Repulsion (MEP) is a tool utilized for analyzing the reactivity of molecules. In the majority of MEP diagrams, the electrophilic attack is depicted in red, while the nucleophilic attack is represented in blue. Additionally, MEP diagrams illustrate the molecular shape, size, and the distribution of negative, positive, and neutral electrostatic potentials, which are indicated through color grading’s. This aids researchers in understanding the physicochemical properties of molecules. The various colors in Fig. 4 correspond to different electrostatic potential values at the surface of the 2-(methylphenyl)-,4-,5-diphenyl-1H-imidazole and its derivatives. The electrostatic potential increases from red to orange, then to yellow, green, and blue. The color coding on the maps was found to be within this specific range of -1.414a.u (deepest red) to 1.414a.u (deepest blue), -1.521a.u (deepest red) to 1.521a.u (deepest blue) and-1298a.u (deepest red) to 1.298a.u (deepest blue) for 2-(2-methylphenyl), 4,5-diphenyl-1H-imidazole, 2-(2-methylphenyl), 4-(4-hydrophenyl), 5-phenyl-1H-imidazole and 2-(2-methylphenyl), 4-(4-nitrophenyl), 5-phenyl-1H-imidazole respectively. In this context, the red color signifies strongest repulsion, whereas the blue color denotes strongest attraction. Areas with negative V(r) values are typically linked to the solitary pair of an electronegative element [48]. Here, the Molecular electrostatic potential surface of 2-(methylphenyl)-,4-,5-diphenyl-1H-imidazole and its derivatives is shown in Figure 4. It illustrates the determined three-dimensional electric potential across the Van der Waals interface. The negative and positive zones linked to regions on the MESP are utilized to examine specific features of the molecular interactions present in the system. The MEPS graphically represent local maxima and minima within the charge distributions on the Van der Waals surface, signifying electronic donor and acceptor sites. The MEPS plot for molecule 2-(methylphenyl)-,4-,5-diphenyl-1H-imidazole shows the effect of the substituents (donor (OH) and accepter (NO2) groups) [34]. Fig. 4 illustrates that there is a negative electrostatic potential surrounding the oxygen atoms in the N-O and H-O groups, while a positive electrostatic potential is observed around the hydrogen atoms and the ring structure. The predominant light green color likely indicates the midpoint between the two extremes, red and blue. This examination offers insights into areas of the compound that are susceptible to intermolecular interactions [48].
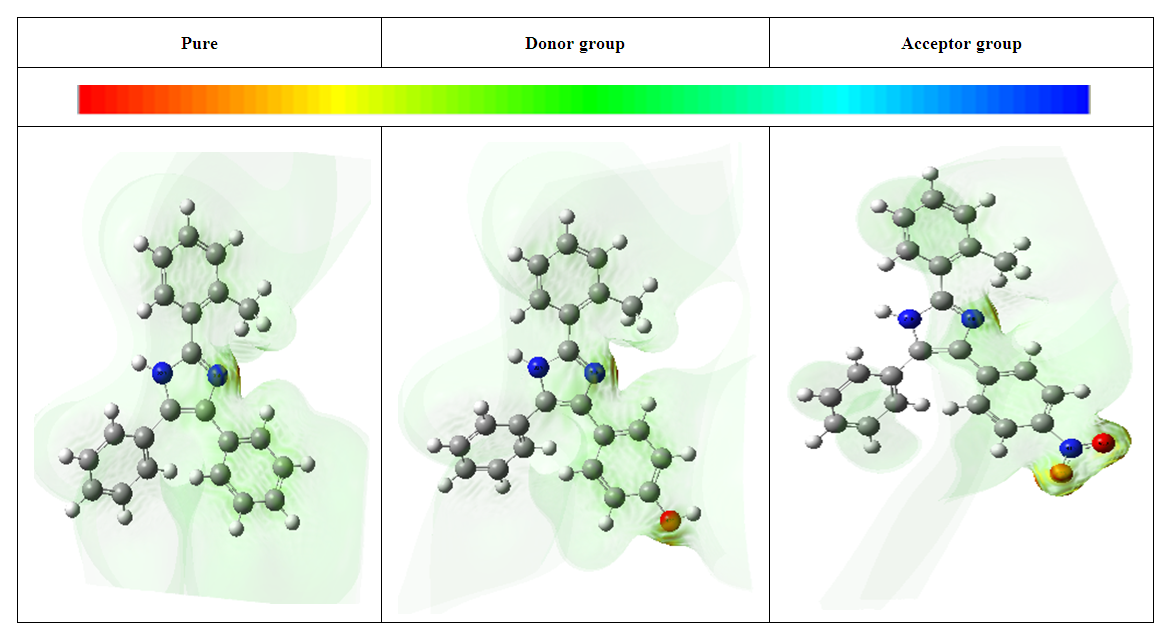 | Figure 4. Molecular electrostatic potential surfaces of 2-(methylphenyl)-,4-,5-diphenyl-1H-imidazole and its derivatives |
4. Conclusions
- In conclusion, this work aimed to investigate the heterocyclic compounds of some substituted (donor (OH) and accepter (NO2) groups) in ortho, meta, and para positions of 2-(2-methylphenyl)-,4-,5-diphenyl-1H-imidazole derivatives. We have performed the DFT/B3LYP calculations to determine the geometric of three molecules in the gas phase. The minimal potential energy distribution (PED) occurred in the para position for derivatives. The molecular structure of molecules was verified through the utilization of IR spectroscopy method. The molecules were subjected to UV-Vis spectral analysis using TD-DFT calculations, which revealed the calculated major singlet-singlet electronic transition for these compounds as HOMO (MO:82) →LUMO (MO:83) at 3.7651eV (329.30nm) with f=0.3962a.u, HOMO (MO:86) →LUMO (MO:87) at 3.6184eV (342.65nm) with f=0.3147a.u and HOMO (MO: 93) →LUMO+1 (MO:95) at 3.9496eV (313.92nm) with f= 0.4598a.u in gas phase are assigned to π→π* transition for original compound with donor (OH) and accepter (NO2) groups respectively. Large values of the energy difference between HOMO and LUMO are 4.19, 4.04, and 3.57 eV, respectively—provide important information regarding the aforementioned compounds. Using the same level theory at 6-311G(d,p) basis sets, the frontier molecular orbital analysis and global reactivity descriptors have been predicted. The chemical hardness values of 2.1, 2.02, and 1.79, which are higher than the chemical softness values of 0.24, 0.12, and 0.14, respectively, lead us to believe that our compounds 2-(2-methylphenyl)-,4-,5-diphenyl-1H-imidazole, 2-(2-methylphenyl)-,4-(4-hydroxyphenyl), 5-phenyl-1H-imidazole, and 2-(2-methylphenyl)-,4-(4-nitrophenyl), 5-phenyl-1H-imidazole are hard molecules. Molecular electrostatic potential surfaces (MEPS) illustrate that the electronegativity increase when the acceptor group (NO2) substituent and decreased decrease when the donor (OH) is substituted.
ACKNOWLEDGEMENTS
- Declares none.
References
| [1] | A. M. Ghaleb and A. Q. Ahmed, “Structural, electronic, and optical properties of sphalerite ZnS compounds calculated using density functional theory (DFT),” Chalcogenide Lett., vol. 19, no. 5, pp. 309–318, May 2022, doi: 10.15251/CL.2022.195.309. |
| [2] | N. Mohammed, S. J. Shakkor, S. M. Abdalhadi, and Y. K. Al-Bayati, “Two multifunctional benzoquinone derivatives as small molecule organic semiconductors for bulk heterojunction and perovskite solar cells,” Main Gr. Chem., vol. 21, no. 4, pp. 943–952, Dec. 2022, doi: 10.3233/MGC-210187. |
| [3] | L. Kang, F. Liang, X. Jiang, Z. Lin, and C. Chen, “First-Principles Design and Simulations Promote the Development of Nonlinear Optical Crystals,” Acc. Chem. Res., vol. 53, no. 1, pp. 209–217, 2020, doi: 10.1021/acs.accounts.9b00448. |
| [4] | H. Wu et al., “Designing Silicates as Deep‐UV Nonlinear Optical (NLO) Materials using Edge‐Sharing Tetrahedra,” Angew. Chemie, vol. 132, no. 23, pp. 9007–9011, 2020, doi: 10.1002/ange.202001855. |
| [5] | S. Chinnasami, M. Manikandan, S. Chandran, R. Paulraj, and P. Ramasamy, “Growth, Hirshfeld surfaces, spectral, quantum chemical calculations, photoconductivity and chemical etching analyses of nonlinear optical p-toluidine p-toluenesulfonate single crystal,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 206, pp. 340–349, 2019, doi: 10.1016/j.saa.2018.08.015. |
| [6] | P. K. M. Lokhande, D. S. Patil, M. M. Kadam, and N. Sekar, “Theoretical Investigation of Optical and Nonlinear Optical (NLO) Properties of 3-Azabenzanthrone Analogues: DFT and TD-DFT Approach.,” ChemistrySelect, vol. 4, no. 34, pp. 10033–10045, 2019, doi: 10.1002/slct.201901681. |
| [7] | S. Sarwar et al., “Deciphering the Role of Alkali Metals (Li, Na, K) Doping for Triggering Nonlinear Optical (NLO) Properties of T-Graphene Quantum Dots: Toward the Development of Giant NLO Response Materials,” ACS Omega, vol. 7, no. 28, pp. 24396–24414, 2022, doi: 10.1021/acsomega.2c01746. |
| [8] | J. Mary Linet and S. Jerome Das, “Optical, mechanical and transport properties of unidirectional grown l-tartaric acid bulk single crystal for non-linear optical application,” Mater. Chem. Phys., vol. 126, no. 3, pp. 886–890, 2011, doi: 10.1016/j.matchemphys.2010.12.020. |
| [9] | J. S. Salem, J. H. Abdulwahid, S. Beebany, and B. L. Mohammed, “Synthesis, Identification, and Antibacterial Effect Assessment of Some New 1, 4-Thiazepines, Derived from Substituted Diphenyl Acrylamides and Diphenyl Dienones,” Chem. Methodol., vol. 7, no. August, pp. 509–523, 2023, doi: 10.22034/CHEMM.2023.392659.1668. |
| [10] | P. Of, “DERIVATIVES USING GREEN Syzygium Cumini SEED,” vol. 15, no. 3, pp. 1861–1866, 2022. |
| [11] | M. Ganga and K. R. Sankaran, “Synthesis, spectral characterization, DFT, docking studies and cytotoxic evaluation of 1-(4-fluorobenzyl)-2,4,5-triphenyl-1H-imidazole derivatives,” Chem. Data Collect., vol. 28, p. 100412, 2020, doi: 10.1016/j.cdc.2020.100412. |
| [12] | S. Hosseini, A. R. Kiasat, and A. Farhadi, “Fe3O4@SiO2/Bipyridinium Nanocomposite as a Magnetic and Recyclable Heterogeneous Catalyst for the Synthesis of Highly Substituted Imidazoles Via Multi-Component Condensation Strategy,” Polycycl. Aromat. Compd., vol. 41, no. 4, pp. 761–771, 2021, doi: 10.1080/10406638.2019.1616306. |
| [13] | O. Ghashghaei, F. Seghetti, and R. Lavilla, “Selectivity in multiple multicomponent reactions: Types and synthetic applications,” Beilstein J. Org. Chem., vol. 15, pp. 521–534, 2019, doi: 10.3762/bjoc.15.46. |
| [14] | N. A. Shekhovtsov, E. B. Nikolaenkova, A. A. Ryadun, D. G. Samsonenko, A. Y. Tikhonov, and M. B. Bushuev, “ESIPT-Capable 4-(2-Hydroxyphenyl)-2-(Pyridin-2-yl)-1H-Imidazoles with Single and Double Proton Transfer: Synthesis, Selective Reduction of the Imidazolic OH Group and Luminescence,” Molecules, vol. 28, no. 4, 2023, doi: 10.3390/molecules28041793. |
| [15] | C. E. Bell, A. Y. Shaw, F. De Moliner, and C. Hulme, “MCRs reshaped into a switchable microwave-assisted protocol toward 5-aminoimidazoles and dihydrotriazines,” Tetrahedron, vol. 70, no. 1, pp. 54–59, 2014, doi: 10.1016/j.tet.2013.11.035. |
| [16] | A. Jayashree, B. Narayana, S. M. Kumar, K. R. Raghi, B. K. Sarojini, and T. K. M. Kumar, “Synthesis, X-ray crystal structure, Hirshfeld surface analysis, DFT, MESP and molecular docking studies of 2-(4-bromophenyl)-1-(3-fluoro-4-methylphenyl)-4,5-diphenyl-1H-imidazole,” Chem. Data Collect., vol. 21, no. May, p. 100237, 2019, doi: 10.1016/j.cdc.2019.100237. |
| [17] | Y. Erdogdu, D. Manimaran, M. T. Güllüoǧlu, M. Amalanathan, I. Hubert Joe, and Ş. Yurdakul, “FT-IR, FT-Raman, NMR spectra and DFT simulations of 4-(4-fluoro-phenyl)-1H- imidazole,” Opt. Spectrosc. (English Transl. Opt. i Spektrosk., vol. 114, no. 4, pp. 525–536, 2013, doi: 10.1134/S0030400X13040073. |
| [18] | N. Fridman, M. Kaftory, Y. Eichen, and S. Speiser, “Crystal structures and solution spectroscopy of lophine derivatives,” J. Mol. Struct., vol. 917, no. 2–3, pp. 101–109, 2009, doi: 10.1016/j.molstruc.2008.07.003. |
| [19] | P. Shojaei, B. Mokhtari, and M. Ghorbanpoor, “Synthesis, in vitro antifungal evaluation and docking studies of novel derivatives of imidazoles and benzimidazoles,” Med. Chem. Res., vol. 28, no. 9, pp. 1359–1367, 2019, doi: 10.1007/s00044-019-02369-7. |
| [20] | G. Șerban Andrei, B. F. Andrei, and P. R. Roxana, “Imidazole Derivatives and their Antibacterial Activity - A Mini-Review,” Mini-Reviews Med. Chem., vol. 21, no. 11, pp. 1380–1392, 2020, doi: 10.2174/1389557520999201209213648. |
| [21] | K. S. Khandare, R. T. Maharaj, L. A. Chate, S. N. Wanjari, M. P. Ambatkar, and P. B. Khedekar, “Design, Synthesis and Biological Evaluation of Some Novel Imidazole Derivatives for Antibacterial Activity,” Anti-inflamm. Antibact. Act. Artic. WORLD J. Pharm. Pharm. Sci., no. September, 2020, doi: 10.20959/wjpps20209-17041. |
| [22] | P. A. Nikitina, N. I. Bormotov, L. N. Shishkina, A. Y. Tikhonov, and V. P. Perevalov, “Synthesis and antiviral activity of 1-hydroxy-2-(2-hydroxyphenyl)imidazoles against vaccinia virus,” Russ. Chem. Bull., vol. 68, no. 3, pp. 634–637, 2019, doi: 10.1007/s11172-019-2467-6. |
| [23] | S. Anthal, B. Narayana, B. K. Sarojini, and R. Kant, “Synthesis, X-ray crystal structure studies and molecular docking analysis of 2-(3,4-dimethoxyphenyl)-4,5-diphenyl-1H-imidazole,” Chem. Data Collect., vol. 15–16, pp. 67–74, 2018, doi: 10.1016/j.cdc.2018.04.004. |
| [24] | Z. W. Li et al., “Synthesis and evaluation of the antitumor activity of novel 1-(4-Substituted phenyl)-2-ethyl imidazole apoptosis inducers in vitro,” Molecules, vol. 25, no. 18, pp. 1–13, 2020, doi: 10.3390/molecules25184293. |
| [25] | S. Chauhan, V. Verma, D. Kumar, and A. Kumar, “Synthesis, antimicrobial evaluation and docking study of triazole containing triaryl-1H-imidazole,” Synth. Commun., vol. 49, no. 11, pp. 1427–1435, 2019, doi: 10.1080/00397911.2019.1600192. |
| [26] | A. A. Marzouk et al., “Design, synthesis and anticonvulsant activity of new imidazolidindione and imidazole derivatives,” Bioorg. Chem., vol. 101, p. 104020, 2020, doi: 10.1016/j.bioorg.2020.104020. |
| [27] | S. Ç. Yavuz, S. Akkoç, and E. Sarıpınar, “The cytotoxic activities of imidazole derivatives prepared from various guanylhydrazone and phenylglyoxal monohydrate,” Synth. Commun., vol. 49, no. 22, pp. 3198–3209, 2019, doi: 10.1080/00397911.2019.1661481. |
| [28] | P. Sharma, C. Larosa, J. Antwi, R. Govindarajan, and K. A. Werbovetz, “Imidazoles as potential anticancer agents: An update on recent studies,” Molecules, vol. 26, no. 14, 2021, doi: 10.3390/molecules26144213. |
| [29] | Y. Liu, Q. Rong, C. Chen, and Y. L. Hu, “Novel and reusable mesoporous silica supported 4-methylbenzenesul-fonate-functionalized ionic liquids for room temperature highly efficient preparation of 2,4,5-triaryl-1H-imidazoles,” J. Mex. Chem. Soc., vol. 65, no. 4, pp. 535–549, 2021, doi: 10.29356/jmcs.v65i4.1529. |
| [30] | J. Jayabharathi, C. Karunakaran, V. Kalaiarasi, and P. Ramanathan, “Donor-acceptor binding interaction of 1-(naphthalene-1-yl)-2,4,5-triphenyl-1H-imidazole with semiconductor nanomaterials,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 137, pp. 333–337, 2015, doi: 10.1016/j.saa.2014.08.048. |
| [31] | S. Naidoo and V. Jeena, “One-Pot, Two-Step Metal and Acid-Free Synthesis of Trisubstituted Imidazole Derivatives via Oxidation of Internal Alkynes Using an Iodine/DMSO System,” European J. Org. Chem., vol. 2019, no. 5, pp. 1107–1113, 2019, doi: 10.1002/ejoc.201801584. |
| [32] | D. Sinha, S. Biswas, M. Das, and A. Ghatak, “An eco-friendly, one pot synthesis of tri-substituted imidazoles in aqueous medium catalyzed by RGO supported Au nano-catalyst and computational studies,” J. Mol. Struct., vol. 1242, p. 130823, 2021, doi: 10.1016/j.molstruc.2021.130823. |
| [33] | K. Anandhan et al., “1H-NMR, Photophysical, and pH Studies of 4-(4,5-Diphenyl-1H-imidazol-2-yl)benzaldehyde through Experimental and DFT Theoretical Analysis,” ChemistrySelect, vol. 5, no. 1, pp. 415–425, 2020, doi: 10.1002/slct.201904505. |
| [34] | A. E. Castillo et al., “Spectroscopic characterization of 4,5-diphenyl-2-(2,4,5-trimethoxyphenyl)-1H-imidazole obtained from the condensation of benzyl. Experimental and DFT approach,” J. Mol. Struct., vol. 1246, p. 131269, 2021, doi: 10.1016/j.molstruc.2021.131269. |
| [35] | S. A. Gandhi, U. H. Patel, R. D. Modh, Y. Naliyapara, and A. S. Patel, “Quantum Chemical Calculations (Ab Initio & DFT), Hirshfeld Surface Analysis, Crystal Structure and Molecular Docking Study of 2-Chloro-4-(4-fluoro-phenyl)-6-isopropyl-pyrimidine-5-carboxylic Acid Methyl Ester,” J. Chem. Crystallogr., vol. 46, no. 10–12, pp. 387–398, 2016, doi: 10.1007/s10870-016-0668-5. |
| [36] | K. Govindarasu, E. Kavitha, and N. Sundaraganesan, “Synthesis, structural, spectral (FTIR, FT-Raman, UV, NMR), NBO and first order hyperpolarizability analysis of N-phenylbenzenesulfonamide by density functional theory,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 133, pp. 417–431, 2014, doi: 10.1016/j.saa.2014.06.040. |
| [37] | H. F. Clausen, M. S. Chevallier, M. A. Spackman, and B. B. Iversen, “Three new co-crystals of hydroquinone: Crystal structures and Hirshfeld surface analysis of intermolecular interactions,” New J. Chem., vol. 34, no. 2, pp. 193–199, 2010, doi: 10.1039/b9nj00463g. |
| [38] | S. Riahi, M. R. Ganjali, A. B. Moghaddam, and P. Norouzi, “Molecular geometry, vibrations and electrode potentials of 2-(4,5-dihydroxy-2-methylphenyl)-2-phenyl-2H-indene-1,3-dione; experimental and theoretical attempts,” J. Mol. Model., vol. 14, no. 4, pp. 325–333, 2008, doi: 10.1007/s00894-008-0273-4. |
| [39] | S. Muthu and E. Isac Paulraj, “Spectroscopic and molecular structure (monomeric and dimeric structure) investigation of 2-[(2-hydroxyphenyl) carbonyloxy] benzoic acid by DFT method: A combined experimental and theoretical study,” J. Mol. Struct., vol. 1038, pp. 145–162, 2013, doi: 10.1016/j.molstruc.2013.01.043. |
| [40] | S. Panchapakesan, K. Subramani, and B. Srinivasan, “Growth, characterization and quantum chemical studies of an organic single crystal: 3-Aminopyridine 4-Nitrophenol for opto-electronic applications,” J. Mater. Sci. Mater. Electron., vol. 28, no. 8, pp. 5754–5775, 2017, doi: 10.1007/s10854-016-6247-x. |
| [41] | M. Karabacak, M. Cinar, and M. Kurt, “DFT based computational study on the molecular conformation, NMR chemical shifts and vibrational transitions for N-(2-methylphenyl) methanesulfonamide and N-(3-methylphenyl) methanesulfonamide,” J. Mol. Struct., vol. 968, no. 1–3, pp. 108–114, 2010, doi: 10.1016/j.molstruc.2010.01.033. |
| [42] | M. Karabacak, M. Çinar, A. Çoruh, and M. Kurt, “Theoretical investigation on the molecular structure, Infrared, Raman and NMR spectra of para-halogen benzenesulfonamides, 4-X-C6H4SO2NH2 (X = Cl, Br or F),” J. Mol. Struct., vol. 919, no. 1–3, pp. 26–33, 2009, doi: 10.1016/j.molstruc.2008.08.007. |
| [43] | S. Chandran, R. Paulraj, and P. Ramasamy, “Structural, optical, thermal, photoconductivity, laser damage threshold and fluorescence analysis of an organic material: β-P-amino benzoic acid single crystal,” Opt. Mater. (Amst)., vol. 52, pp. 49–55, 2016, doi: 10.1016/j.optmat.2015.11.044. |
| [44] | K. Sambathkumar, S. Jeyavijayan, and M. Arivazhagan, “Electronic structure investigations of 4-aminophthal hydrazide by UV-visible, NMR spectral studies and HOMO-LUMO analysis by ab initio and DFT calculations,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 147, pp. 124–138, 2015, doi: 10.1016/j.saa.2015.03.012. |
| [45] | M. Prabhaharan, A. R. Prabakaran, S. Gunasekaran, and S. Srinivasan, “DFT studies on vibrational spectra, HOMO-LUMO, NBO and thermodynamic function analysis of cyanuric fluoride,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 136, no. PB, pp. 494–503, 2015, doi: 10.1016/j.saa.2014.09.062. |
| [46] | S. Muthu, E. Elamurugu Porchelvi, M. Karabacak, A. M. Asiri, and S. S. Swathi, “Synthesis, structure, spectroscopic studies (FT-IR, FT-Raman and UV), normal coordinate, NBO and NLO analysis of salicylaldehyde p-chlorophenylthiosemicarbazone,” J. Mol. Struct., vol. 1081, pp. 400–412, 2015, doi: 10.1016/j.molstruc.2014.10.024. |
| [47] | N. Uludağ and G. Serdaroğlu, “An improved synthesis, spectroscopic (FT-IR, NMR) study and DFT computational analysis (IR, NMR, UV–Vis, MEP diagrams, NBO, NLO, FMO) of the 1,5-methanoazocino[4,3-b]indole core structure,” J. Mol. Struct., vol. 1155, no. November 2017, pp. 548–560, 2018, doi: 10.1016/j.molstruc.2017.11.032. |
| [48] | M. S. Almutairi et al., “Spectroscopic (FT-IR, FT-Raman, UV, 1H and 13C NMR) profiling and computational studies on methyl 5-methoxy-1H-indole-2-carboxylate: A potential precursor to biologically active molecules,” J. Mol. Struct., vol. 1133, no. 2017, pp. 199–210, 2017, doi: 10.1016/j.molstruc.2016.12.004. |
| [49] | B. Edwin, M. Amalanathan, and I. Hubert Joe, “Vibrational spectra and natural bond orbital analysis of organic crystal l-prolinium picrate,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 96, pp. 10–17, 2012, doi: 10.1016/j.saa.2012.04.062. |
| [50] | K. R. Ansari and M. A. Quraishi, “Experimental and computational studies of naphthyridine derivatives as corrosion inhibitor for N80 steel in 15% hydrochloric acid,” Phys. E Low-Dimensional Syst. Nanostructures, vol. 69, pp. 322–331, 2015, doi: 10.1016/j.physe.2015.01.017. |
| [51] | M. Karnan, V. Balachandran, and M. Murugan, “Vibrational spectroscopic (FT-IR and FT-Raman) studies, natural bond orbital analysis and molecular electrostatic potential surface of 3-hydroxy-6-methyl-2-nitropyridine,” Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., vol. 96, pp. 51–62, 2012, doi: 10.1016/j.saa.2012.05.007. |
| [52] | T. Ghailane et al., “Experimental and theoretical studies for mild steel corrosion inhibition in 1M HCl by two new benzothiazine derivatives,” Corros. Sci., vol. 76, pp. 317–324, 2013, doi: 10.1016/j.corsci.2013.06.052. |