-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Condensed Matter Physics
p-ISSN: 2163-1115 e-ISSN: 2163-1123
2019; 9(1): 1-5
doi:10.5923/j.ajcmp.20190901.01

First Principle Study of Structural, Elastic and Electronic Properties of Hexagonal Boron Nitride (hex-BN) Single Layer
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAlhassan Shuaibu1, Owolabi Joshua Adeyemi2, Ugbe Raphael Ushiekpan2, Olawale Gabriel Olowomofe3, Bamikole Johnson Akinade4, Odelami Abiodun Kafayat5
1Department of Physics, Faculty of Science, Kaduna State University, Kaduna, Nigeria
2Department of Physics Nigerian Defence Academy, Kaduna, Nigeria
3Department of Physics Ekiti State University, Ado Ekiti, Ekiti State, Nigeria
4Department of Physics, Federal University Lafia, Lafia, Nigeria
5Department of Physics ShehuIdris College of Health Science & Technology Makarfi, Kaduna, Nigeria
Correspondence to: Alhassan Shuaibu, Department of Physics, Faculty of Science, Kaduna State University, Kaduna, Nigeria.
| Email: |  |
Copyright © 2019 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

In this work, the structural and electronic properties of hexagonal boron nitride sheet have been calculated within the density functional theory as implemented in Quantum ESPRESSO (QE) code while the elastic properties have been calculated using Elastic code, which is able to calculate the full second-order elastic stiffness tensor for any crystal structure from ab initio total-energy and/or stress calculations. From our results, The obtained projected density of states (PDOS) shows that the covalent bonding of boron (B), nitrogen (N) is mainly contributed by s,d like-orbitals of B and partially occupied by the 2p like-orbital of N order. From the electronic band structure it is clearly shown that hexagonal boron nitride is narrow band with the semiconductor half metallic nature. The obtained Young’s modulus and shear modulus shows an excellent agreement when compared with available theoretical and experimental data.
Keywords: Hexagonal Boron Nitride, Density Functional Theory, Quantum Espresso, Elastic and electronic properties
Cite this paper: Alhassan Shuaibu, Owolabi Joshua Adeyemi, Ugbe Raphael Ushiekpan, Olawale Gabriel Olowomofe, Bamikole Johnson Akinade, Odelami Abiodun Kafayat, First Principle Study of Structural, Elastic and Electronic Properties of Hexagonal Boron Nitride (hex-BN) Single Layer, American Journal of Condensed Matter Physics, Vol. 9 No. 1, 2019, pp. 1-5. doi: 10.5923/j.ajcmp.20190901.01.
1. Introduction
- In recent years, the ‘wonder material’ graphene has been explored intensively due to its ultrahigh room-temperature carrier mobility [1, 2] and is touted to be the next-generation electronic material to replace silicon, since silicon is approaching its performance limits.Graphene, the well-publicized and famous two-dimensionalboron nitride (BN) allotrope, have attracted intensive research interest due to their fascinating electronic properties and extensive applications [3, 4]. Among them, Hexagonal boron nitride (h-BN) is an insulator with a large band gap [5, 6] a two-dimensional monolayer of carbon atoms arranged in a honeycomb lattice, it is the building block for other graphitic materials including one-dimensional carbon nanotubes and three-dimensional graphite. It has been theoretically investigated by several scientist for more than sixty years. For instants, Wallace calculated the electronic band structure of graphene using a tight-binding method in 1947, showing its semi-metallic nature and linear energy dispersion around the Fermi energy. [7].Geim and Novoselov of Manchester University demonstrated that graphene could be isolated by using a remarkably simple mechanical exfoliation technique which is commonly called the Scotch tape methodin 2004 [8].In 2005, the Manchester group [9] and Philip Kim’s group at Columbia University independently observed the quantum Hall effect in grapheme [10]. This shows the quantum Hall plateaus were found to be quantized in half-integer values which provided convincing evidence of the two-dimensional massless chiral nature of charge carriers in this system.These early discoveries have initiated intense research activities worldwide on graphene and other two-dimensional materials. The exploding interests in graphene have been largely motivated by its remarkable electronic properties and its stable form. Due to the linear energy dispersion around the Fermi energy, the low-energy charge carriers in graphene has described by relativistic Dirac equation rather than the Schrodinger equation. However, the high mobility of charge carriers and the ambipolar transport characteristics make graphene particularly promising for high frequency applications. It was demonstrated by Lin and his group that high frequency operation, at 100 GHz, was possible in graphene-based transistors [11] meaning that it can be integrated into a huge number of applications. The only problem with graphene is that high-quality graphene is a great conductor that does not have a band gap (a semi-metal with a zero band gap). Due to this intrinsic property, it is not possible to switch off the devices with graphene as the channel material, resulting in very poor ON/OFF ratio. Although a band gap can be introduced into graphene, but the process is extremely complex to make and will result in a loss of mobility.In order to realize graphene’s full potential as a post-silicon electronic material, a sizable bandgap must be created [12]. Although a small bandgap can be engineered in nano-ribbons and bilayer graphene under strong electric field, but large gap has not been created at this stage sufficiently.Mechanical properties of graphene are also extraordinary due to the strong carbon-carbon covalent bonds. The tensile strength of graphene is measured to be about 300 times greater than steel [13]. In addition, graphene can sustain a large degree of stretching and bending without structural damage. These properties together with the optical transparency make graphene very attractive in potential applications in solar cells, touch screen displays, and flexible optoelectronic devices. The major focus of our research is to obtain the structural properties, electronic properties and elastic properties of BN for its potential applications.
2. Computational Method
- All the calculations reported in this work were performed using the Quantum-ESPRESSO code [14] with the version of 5.1 which is based on density-functional theory (DFT) in which plane wave basis sets for expansion of the electronic wave function and conjugated gradient electronic minimization were used. In this package, electron-ion interactions were described using the norm-conserving or ultrasoftpseudopotentials. The exchange and correlation energies were calculated with the Perdew-Burke-Ernzerhof (PBE) form of the generalized gradient approximation (GGA) [15, 16]. Geometry optimizations were performed by the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm. The k-points, lattice constant and plane wave cutoff energy were thoroughly tested. In this study a k-point grid of 4x4x4 were used for the bulk properties. The optimized value for plane wave cutoff of 37 Ry used for all calculations. All the known cell parameters were fixed at the experimental value of the crystal during geometry optimizations while all the atom positions were minimized with symmetries. Where no experimental value were available the atom-atom distance was approximated by adding the covalent radii of the elements [17].For the pseudopotential generation Brillouin Zone integrations were performed using a 8×8×1 Monkhorst and Pack special point grids using Gaussian smearing technique with a smearing width of 0.02 Ry in order to smooth the Fermi distribution. The Kohn-Sham orbitals are expanded in a plane wave basis setby obtaining the structural stability and electronic properties such as (ground state total energy, density of state (DOS) projected density of state (PDOS), magnetic moments and charge density distribution) within the DFT as implemented on the Quantum ESPRESSO package. While dealing with the PW basis the first step was to have a details of the crystal structure for the compound of interest, therefore, our structural parameters was obtained from available literatures specifically experimental database reported in [18, 19]. Then we optimized them (relaxation calculation) using DFT-GGA by applying the Broyden-Fletcher-Goldfarb-Shannon (BFGS) algorithm [20], unless otherwaise the optimized parameters close to the experimental values was taking and employed in our calculations.In a crystal structural optimization, one has to look for the structure with a minimum potential energy surface (PES) that is closest to the starting configuration. In practice one has to search for the ionic configuration for which all the forces are zero. A Quasi-Newton ionic relaxation within the Broyden Fletcher Goldfarb Shanno algorithm has been one of the most successful approximations within a theoretical framework for more than a decade. In order to determine the elastic properties we consider the general deformation state, such that the number of independent components of the second, third, fourth and fifth order elastic tensors are 23, 58, 136, and 272 respectively. Conversely, there are only fourteen independent elastic constants need to be explicitly considered due to the symmetries of the atomic lattice point group of h-BN which consists of a six-fold rotational axis and six mirror planes.The fourteen independent elastic constants of h-BN are determined by a least-squares fit to the stress-strain results from DFT based first-principles studies in two steps as implemented in Elastic code of the QE [21]. In the first step, we use a least-squares fit of five stress-strain responses.Five relationships between stress and strain are necessary because there are five independent fifth-order elastic constants. We obtain the stress-strain relationships by simulating the following deformation states: uni-axial strain in the zigzag direction (zigzag); uni-axial strain in the armchair direction (armchair); and equibiaxial strain (biaxial). From the first step, the components of the second-order elastic constants, the third-order elastic constants, and the fourth order elastic constants are over-determined (i.e, the number of linearly independent variables are greater than the number of constrains), and the fifth-order elastic constants are well-determined (the number of linearly independent variables are equal to the number of constrains). Under such circumstances, the second step is needed: least-square solution to these over- and well- determined linear equations.
3. Results and Discussions
- A Structural and Elastic PropertiesI. Atomic structureWe first optimize the equilibrium lattice constant for h-BN. The total energy as a function of lattice spacing is obtained by specifying nine lattice constants varying from 3.3 Å to 4.1 Å, with full relaxations of all the atoms. A least-square fit of the energies versus lattice constants with a fourth-order polynomial function yields the equilibrium lattice constant as a = 3.731 Å. The most energetically favorable structure is set as the strain-free structure in this study and the atomic structure, as well as the conventional cell is shown in Fig. 1. Specifically, the bond length of B-N bond is 2.154 Å. The B-N-B and B-N-B angles are 120 and all atoms are within one plane. Our result of bond length is in good agreement with previous DFT calculations of A-BNNR sheet [22].
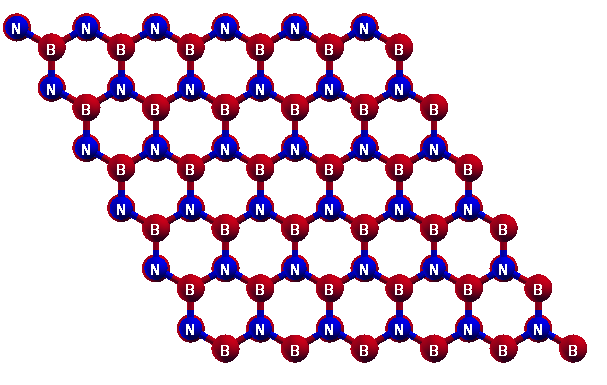 | Figure 1. Optimized Hexagonal Boron Nitride structure |
 | Figure 2. Calculated X-Ray of Hexagonal Boron Nitride |
|
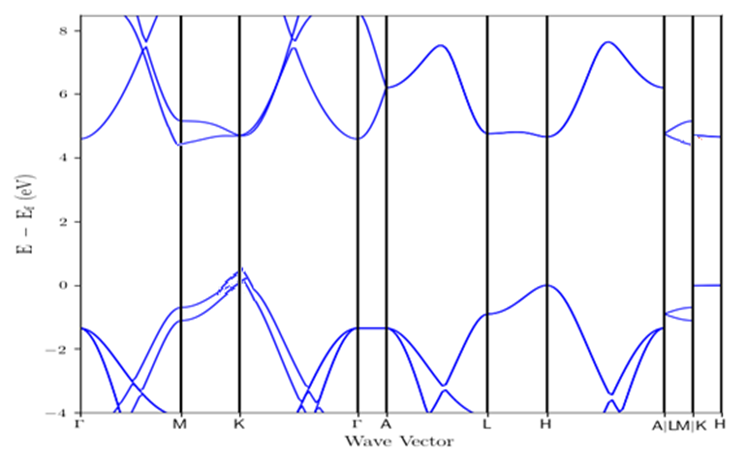 | Figure 3. Calculated Band Structure of Hexagonal Boron Nitride |
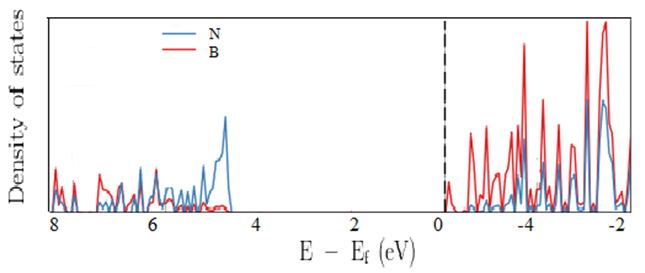 | Figure 4. Calculated Density of State of H-BN |
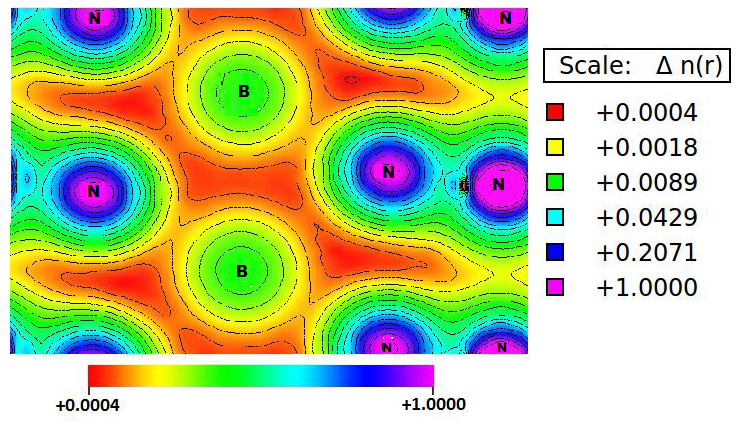 | Figure 5. The top view of the calculated chargedensity plot of the h-BN ssystem with colors scale n(r) indicating ranges of charge accumulation and depletionina.u |
4. Conclusions
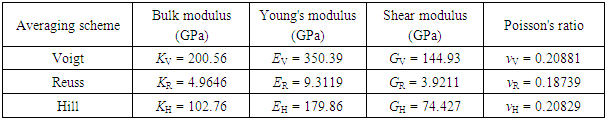
- In summary, we studied the structural, electronic and elastic properties of h-BN under various strains using DFT based first principles calculations. It is observed that h-BN shows a nonlinear elastic deformation up to a definitive strain, which is 0.20881, 0.18739, and 0.20829 for Voigt, Reuss, and Hill respectively, which are large Poisson ratio compared to hexagonal graphene. We also found that the h-BN can sustain much smaller strains before the rupture. The nonlinear elasticity of h-BN was investigated. We found an accurate continuum description of the elastic properties of h-BN by explicitly determining the fourteen independent components of high order (up to fifth order) elastic constants from the fitting of the stress-strain curves obtained from DFT calculations. This data is useful to develop a continuum description which is suitable for incorporation into a finite element analysis model for its applications in large scale. The second order elastic constants including in-plane stiffness are predicted to monotonically increase with pressure while Poisson’s ratio monotonically decreases with increasing pressure.