-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Condensed Matter Physics
p-ISSN: 2163-1115 e-ISSN: 2163-1123
2014; 4(1): 13-19
doi:10.5923/j.ajcmp.20140401.02
Ab-initio Calculations of Structural, Electronic, Optical,  Dynamic and Thermodynamic Properties of HgTe and HgSe
Dynamic and Thermodynamic Properties of HgTe and HgSe
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLM. N. Secuk, M. Aycibin, B. Erdinc, S. E. Gulebaglan, E. K. Dogan, H. Akkus
Department of Physics, Yuzuncu Yil University, Van, 65080, Turkey
Correspondence to: M. N. Secuk, Department of Physics, Yuzuncu Yil University, Van, 65080, Turkey.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
In present work, using the density functional theory within local density approximation, the structural optimization, electronic band structure, density of electron states, optical, dynamic and thermodynamic properties of HgTe and HgSe were investigated. It was found that HgTe and HgSe show semiconducting property with a direct band gap at high symmetry point gamma with the value of 0.99 eV since conduction band does not interact with uppermost state of valance band because of existence of heavy holes. Real and imaginary parts of dielectric function as a function of photon energy were studied and photon wavelength dependence of refractive index was compared with experimental result. Also, temperature dependent thermodynamic properties such as Helmholtz free energy, internal energy, entropy and specific heat have been worked. The calculated and experimental results are in a good agreement.
Keywords: Density Functional Theory, Electronic Structure, Optical Properties, Dynamic Properties
Cite this paper: M. N. Secuk, M. Aycibin, B. Erdinc, S. E. Gulebaglan, E. K. Dogan, H. Akkus, Ab-initio Calculations of Structural, Electronic, Optical, Dynamic and Thermodynamic Properties of HgTe and HgSe, American Journal of Condensed Matter Physics, Vol. 4 No. 1, 2014, pp. 13-19. doi: 10.5923/j.ajcmp.20140401.02.
Article Outline
1. Introduction
- Many semiconductors of AIIBVI type crystallize in the form of zinc-blende structure. Among those HgTe and HgSe are special because of their different band end structure[1-3]. Mercury chalcogenides are to be used in optoelectronic and spintronic applications[1,4]. Besides, conduction bands of HgTe and HgSe are non-parabolic and can be well explained by Kane model[5]. The cubic HgTe and HgSe are technologically interesting materials with applications in quantum electronics[6]. Compared to other II-IV group semiconductors, few experimental and theoretical studies on these compounds have been performed[7-10]. Delin and Klüner, using an all-electron full-potential linear muffin-tin orbital method, found that in the zinc-blende structure both HgSe and HgTe are semimetals[6]. Arora and Ahujawe reported energy bands, density of states (DOS) and band gaps of these chalcogenides via using Hartree–Fock and density functional theory[11]. Penna et al. calculated the electronic structure of the binary compounds CdTe and HgTe with density functional theory local density approximation (DFT-LDA), empirical pseudopotential method (EPM) and a new full-Brillouin zone (FBZ) k.p method[12]. Rajput and Browne studied lattice dynamics of II-VI compounds by the adiabatic bond charge model[13]. Cordona et al. studied the electronic band structure and the phonon dispersion relations of the zinc-blende (ZB) type mercurychalcogenides[9]. Thermal properties of mercury chalcogenides were also calculated[10].There is no comprehensive study about HgTe and HgSe by using density functional theory as far as we know. So we investigated structural and volume optimization, electronic band structure, density of states, optical properties, dynamic and thermodynamic properties for these compounds.
2. Computational Methods
- The physical properties of HgTe and HgSe crystals were investigated using ABINIT[14] code within the local density approximation based on the density functional theory. The self-consistent pseudopotentials, generated by FHI98PP code[15] with the Ceperley-Alder Perdew-Wang scheme[16], were used for all ab initio calculations. The conjugate gradient minimization method[17] was employed in order to solve the Kohn-Sham equations[18]. The exchange- correlation effects were taken into account within the Ceperley-Alder Perdew-Wang local density functional pseudopotentials (CAPW-LDA-1992)[19]. Plane waves were used as the basis set for the electronic wave functions. For mercury atom the 5d and 6s electrons, for tellurium atom the 5s and 5p electrons and for selenium atom the 4s and 4p electrons were considered as the true valence. First of all, in all calculations total energy of this materials were optimized with respect to cutoff energy and Monkhorst-Pack mesh grid. The obtained total energy calculations were done to a good convergence at 25 Hartree of cutoff energy and 60 k points using 8x8x8 Monkhorst-Pack mesh grid[20] in HgTe and HgSe crystals for structural optimization and electronic band structure. The Brillouin zones of these compounds have been sampled with a 8x8x8 Monkhorst-Pack mesh grid with 60 k points for the calculations of dynamic and thermodynamic properties and a 10x10x10 Monkhorst-Pack mesh grid 110 k points for the calculations of optical properties.
3. Structural Properties
- All the calculations of HgTe and HgSe crystals involve two atoms per face centered cubic unit cell and they have space group symmetry with space group number 216. Firstly, total energy optimization with respect to cutoff energy was performed and value of cutoff energy was calculated as 25 Hartree (Ha) for both compounds as seen in figure 1. Secondly number of k points was optimized by total energy and was calculated as 60 for both HgTe and HgSe as shown in figures 2a and 2b. One of the two atoms (Hg) was placed at the 0.0, 0.0, 0.0 while the other (Te/Se) at 0.5, 0.5, 0.5.Thirdly, lattice and volume parameter optimization were performed according to total energy. Volume parameters were calculated as 447.739 and 424.484 of cubic Bohr respectively as in figure 3. The calculated values of lattice parameters for HgTe and HgSe compounds are 12.144 and 11.556 Bohr, while experimental ones are 12.21 and 11.49 Bohr respectively. As seen, there is a good agreement between calculated and experimental results for lattice parameters. Then, calculated results were used for calculation of all physical properties of these compounds. Total energy was investigated as a function of volume in figure 3 and as a function of pressure of unit cell for HgTe and HgSe crystals as seen in figure 4. It is seen from figures 3 and 4 that minimum values total energies are -67.62 and -68.875 Ha for these compounds. Figure 5 shows pressure as a function of volume of unit cell for HgTe and HgSe crystals.
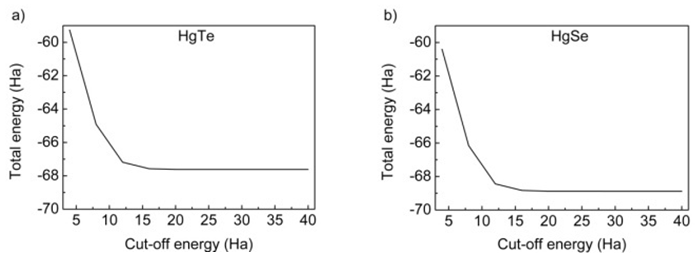 | Figure 1. Cut-off energy-total energy optimization for a) HgTe and b) HgSe |
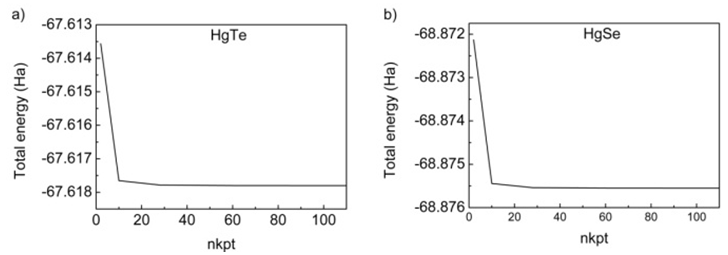 | Figure 2. Number of k points (nkpt) - calculated total energy optimization for a) HgTe and b) HgSe |
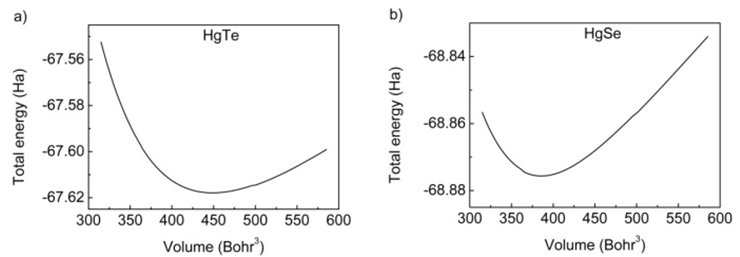 | Figure 3. Total energy-volume dependence for unit cells of HgTe and HgSe |
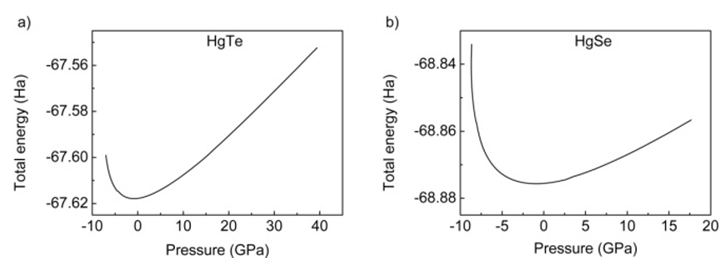 | Figure 4. Total energy-pressure dependence for unit cells of HgTe and HgSe |
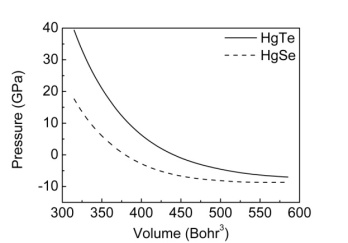 | Figure 5. Pressure-volume dependence for unit cells of HgTe and HgSe |
4. Electronic Properties
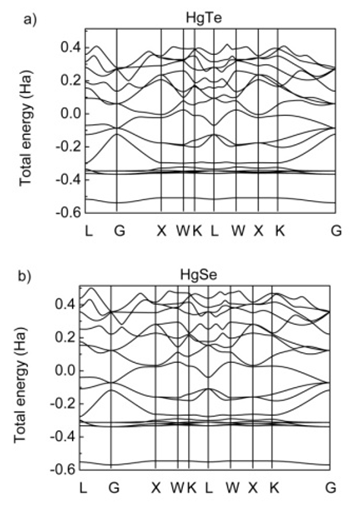 | Figure 6. Electronic band structure for a) HgTe and b) HgSe |
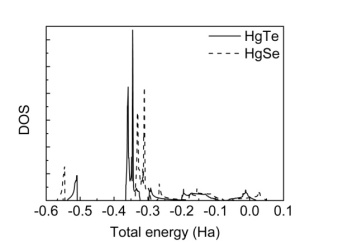 | Figure 7. DOS for HgTe and HgSe |
5. Optical Properties
- HgTe and HgSe crystals are optic crystals and they exhibit the symmetry of the point group 216. The calculated real (ε1) and imaginary (ε2) parts of these compounds are given in figure 8. As seen from these figures, the real part of linear dielectric function, ɛ1, goes to its maximum value at 0 eV of photon energy for both HgTe and HgSe. Furthermore, one can see from figure 8 that the static dielectric constants for HgTe and HgSe crystals are 15.0 and 13.5, respectively. The main peak values in the imaginary parts of linear dielectric functions calculated in present work are at 1.9, 4 and 5.7 eV for HgTe and 2.0, 5.0 and 6.6 eV for HgSe. The energy loss functions (L), for both volume and surface, are presented in figures 9 for the compounds HgTe and HgSe. The maximum of calculated surface is at 10 eV, while maxima for volume loss functions are at 14, 18.8, 23.5, 27 and 32 eV for HgTe. On the other hand, maxima of calculated surface are at 11, 13, 16 and 20 eV, while for volume loss functions are at 12, 14.5, 16.5, 20, 21.6, 23.6, 26.4 and 34.6 eV for HgSe. The calculated extinction coefficients (k), absorption coefficients (α), and reflectivities (R) for HgTe and HgSe are shown in figure 10a, 10b and 10c, respectively.
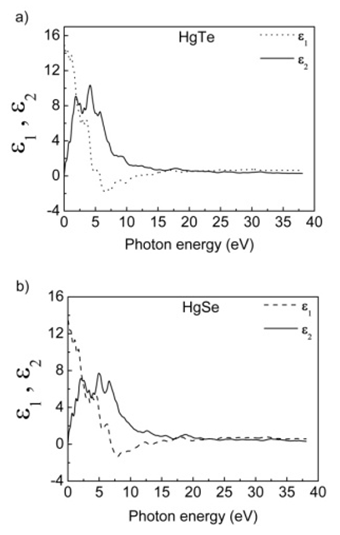 | Figure 8. Real and imaginary parts of dielectric function for a) HgTe and b) HgSe |
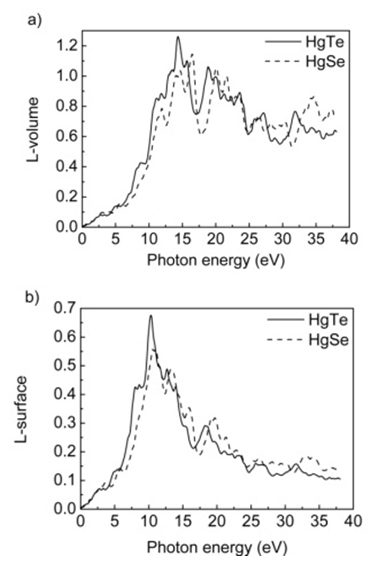 | Figure 9. Loss functions of HgTe and HgSe as a function of photon energy for a) volume and b) surface |
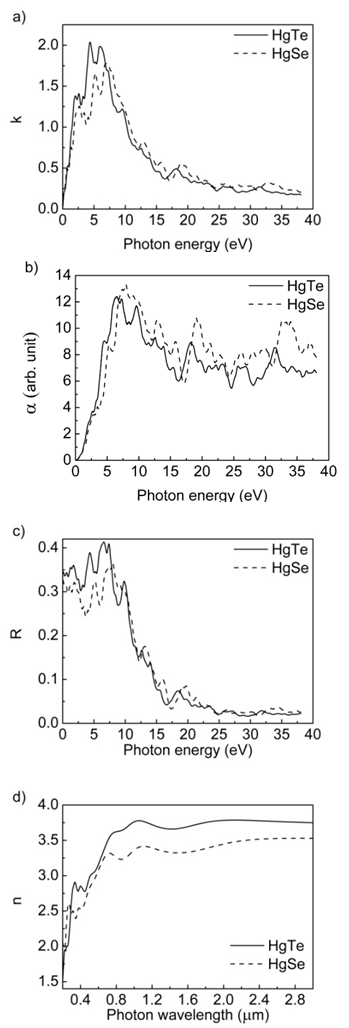 | Figure 10. The calculated a) extinction coefficients, b) absorption coefficients and c) reflectivities as a function of photon energy, d) spectral dependence of refractive index (n) on wavelength |
6. Dynamic and Thermodynamic Properties
- In mercury tellurium and mercury selenide crystals, unit cell contains two atoms at zero pressure. In the end of crystallization of HgTe and HgSe at zero pressure, while the Hg atom is located at the point of (0.0, 0.0, 0.0), Te/Se atom is located at the point of (0.5, 0.5, 0.5), in the reduced coordinates, inside the unit cell. Figure 11 and 12 show the phonon band structures and phonon density of states for these crystals.
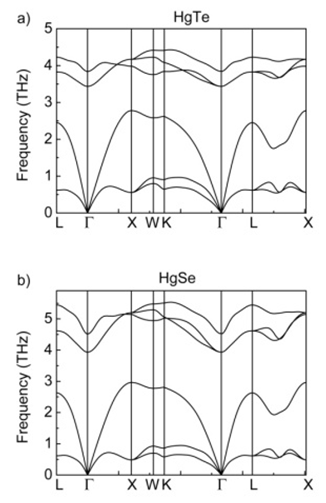 | Figure 11. Phonon band structures for a) HgTe and b) HgSe |
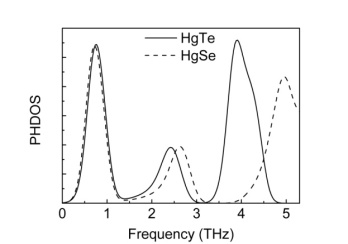 | Figure 12. Phonon density of states for HgTe and HgSe |
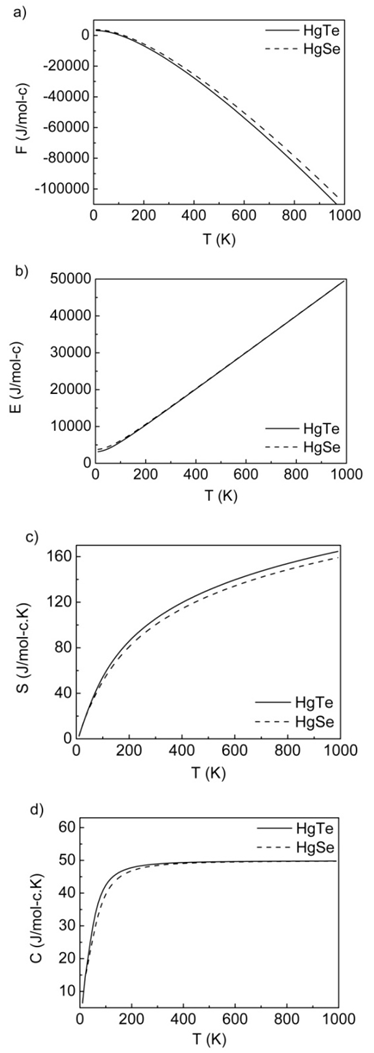 | Figure 13. a) Helmholtz free energy, b) internal energy, c) entropy and d) constant volume specific heat for HgTe and HgSe as a function of temperature |
7. Conclusions
- Structural and volume optimization, electronic band structure, density of states, optical properties, dynamic and thermodynamic properties of HgTe and HgSe were investigated by using ABINIT[14] code within the local density approximation based on the density functional theory. HgTe and HgSe show semiconducting property with a direct band gap at high symmetry point gamma with the value of 0.99 eV.
ACKNOWLEDGEMENTS
- This work has been supported by The Unit of Scientific Research Projects of Yuzuncu Yil University under project No. 2011-FED-B010.