-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Condensed Matter Physics
p-ISSN: 2163-1115 e-ISSN: 2163-1123
2012; 2(4): 83-87
doi: 10.5923/j.ajcmp.20120204.02
Electronic Structure of AlAs Nanocrystals
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLHaider I. Isa , Ahmed M. Abdul-Lettif
Physics Department, College of Science, University of Babylon, Hilla, Iraq
Correspondence to: Ahmed M. Abdul-Lettif , Physics Department, College of Science, University of Babylon, Hilla, Iraq.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Ab initio restricted Hartree-Fock method within the framework of large unit cell formalism is used to investigate the electronic structure of the core and oxidized surface of AlAs nanocrystals. Large unit cells of 8, 16, 54, 64 and 128 atoms are used in the present analysis. Calculations are performed utilizing the Gaussian (03) software. The investigated properties include the cohesive energy, energy gap, valence bandwidth, conduction bandwidth and the density of states of the energy levels. Interesting results are obtained which reveal that the electronic structure of AlAs nanocrystals differs significantly from that of the bulk AlAs crystal. Also, it is found that the energy gap, valence band width and cohesive energy (absolute value) increase as the AlAs large unit cell size increases, for the core part. Whereas, the energy gap of oxygenated (001) - (1×1) surface decreases with increasing the large unit cell size. The energy gap is controlled by the surface part of the nanocrystal. The surface part has lower symmetry than the core part with smaller energy gap and wider valence bandwidth. The density of states of the core part is higher than that of the surface part. This is due to the broken bonds and the discontinuity at the surface and the existence of new kind of atoms (oxygen atoms). The present method has threefold results: the method can be used to obtain the converged electronic structure of bulk, surface, and nanocrystals.
Keywords: Nanocrystals, Band Structure, Semiconductors, Ab initio, Hartree-Fock , Density of States
1. Introduction
- Aluminium arsenide (AlAs) is a compound semiconductor with an indirect band gap and it has a zinc-blend (zb) structure[1]. AlAs is the most technologically important compound semiconductor material, where it has many optoelectronics and photovoltaic applications in space solar cells, telecommunication and data communication networks, mobiles, telephones, laptop computers, compact disc players, etc[2]. Therefore, aluminium arsenide has been intensively investigated in the recent years, where Bouarissa and Boucenna[3] studied the electronic, optical and mechanical properties of AlAs and they showed that the results are in agreement with the available experimental and theoretical data. Chimata[4] studied theoretically the electronic structure of aluminium arsenide crystal, and they found that the band gap value obtained is in good agreement with the experimental value. Shimazaki and Asai[5] studied the energy band structure of Si, AlP, AlAs, GaP and GaAs using screened Hartree–Fock exchange method, and they found that the direct and indirect band gaps don't agree with experimental values.The aforementioned researches and most of other researches deal with bulk AlAs crystals. Due to our extended literature survey, it is found that the electronic properties of AlAs nanocrystals are not investigated comprehensively. Sothe aim of the present work is to achieve this task. In the present work we introduce the ab-initio restricted Hartree-Fock(RHF) formalism within the frame of large unit cell (LUC) method to investigate the electronic structure of AlAs nanocrystals (ncs) employing Gaussian (03) software[6] to perform the calculations. This method has been chosen in the present work rather than other methods because this method is very reliable to analyse the electronic properties of solids, and because of the significant advantage of ab-initio methods which is the ability to study reactions that involve breaking or formation of covalent bonds, which correspond to multiple electronic states. The downside of ab-initio methods is their computational cost. They often take enormous amounts of computer time, memory and disk space[7].
2. Theory
- The main idea of the large unit cell is computing the electronic structure of the unit cell extended in a special manner at k=0 in the reduced Brillouin zone[8, 9]. Using the linear combination of atomic orbitals (LCAO), the crystal wavefunction in the (LUC-RHF) formalism is written in the following form[10, 11]:
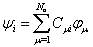 | (1) |
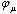 is the atomic orbital, N0 is the number of the atomic orbitals. The total electronic energy can be written as[12, 13, 14]:
is the atomic orbital, N0 is the number of the atomic orbitals. The total electronic energy can be written as[12, 13, 14]: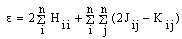 | (2) |
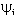 ), Jij and Kij are the Coulomb and exchange operators respectively, and they are expressed as follows:
), Jij and Kij are the Coulomb and exchange operators respectively, and they are expressed as follows: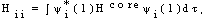 | (3) |
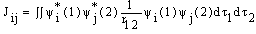 | (4) |
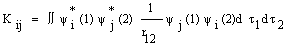 | (5) |
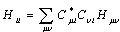 | (6) |
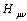 is the core Hamiltonian matrix elements,
is the core Hamiltonian matrix elements,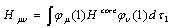 | (7) |
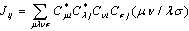 | (8) |
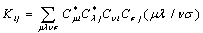 | (9) |
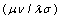 is the differential overlap matrix elements,
is the differential overlap matrix elements,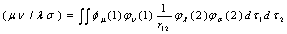 | (10) |
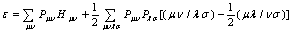 | (11) |
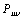 is the density matrix elements,
is the density matrix elements,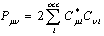 | (12) |
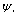 can now be given as:
can now be given as: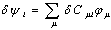 | (13) |
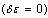 is applied, one can obtain the following final form[15,16]:
is applied, one can obtain the following final form[15,16]: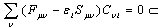 | (14) |
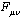 is the Fock matrix,
is the Fock matrix,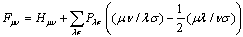 | (15) |
3. Results and Discussion
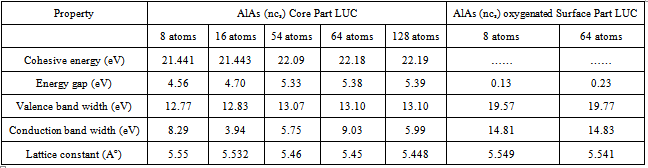
- In the present work we have used Gaussian (3) software to calculate the lattice constant, energy gap, cohesive energy, valence band width and degeneracy of states of AlAs ncs (core and surface). These properties are illustrated in table (1) and they are compared with other experimental and theoretical results in table (2). We divided the work into two parts, core and surface parts. In the core part we chose the number of atoms (8, 16, 54, 64, 128) for the (LUC), while in the surface part the choice of number of atoms is (8 and 64) because these numbers of atoms give cubic shapes.The calculated total energy of AlAs ncs as a function of lattice constant for the LUC of (8) atoms is shown in Fig. (1). The equilibrium lattice constant is at the minimum of the total energy curve.
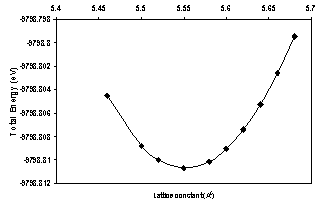 | Figure 1. Total energy as a function of lattice constant for 8 atom (core) LUC |
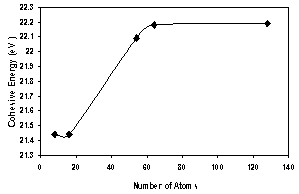 | Figure 2. Cohesive energy as a function of number of atoms for AlAs (ncs) |
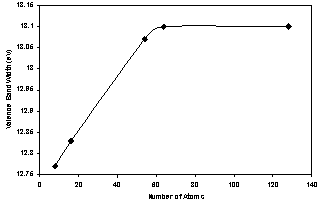 | Figure 3. The valence band width as a function of number of atoms for AlAs (ncs) |
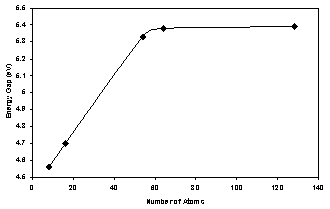 | Figure 4. Energy gap as a function of number of atoms for AlAs( ncs) |
|
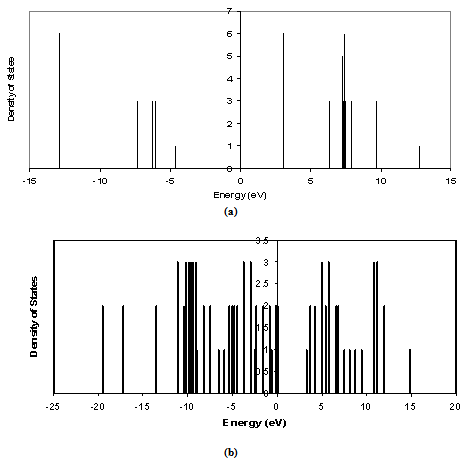 | Figure 5. Density of states as a function of energy levels for 8 atoms LUC: (a) core, and (b) oxygenated (001)-(1×1) slab |
4. Conclusions
- 1. The present work reveals an interesting fact that the electronic structure of AlAs nanocrystals differs significantly from that of the bulk AlAs crystal.2. The present method cannot be used only to investigate the electronic properties of bulk crystals, but it may be also used to investigate the electronic properties of nanocrystals.3. The lattice constant of all size of AlAs ncs (core and surface) decreases with increasing the number of atoms in the LUC.4. The present calculations show that the energy gap and valence band (absolute value) increase as the AlAs (ncs) LUC size increases for both the core and oxygenated (001)-(1×1) surface. Also the cohesive energy (absolute value) for the core part increase with increasing the number of atoms.5. The energy gap is controlled by the surface part of the nanocrystal. The surface part has lower symmetry than the core part with smaller energy gap and wider valence band. The density of states of the core part is higher than that of the surface part. This reflects the high symmetry and equal bond lengths and angles in the perfect AlAs nanocrystals structure.
References
| [1] | Grundmann M.,: 'The Physics of Semiconductors' (Springer-Verlag Berlin Heidelberg, 2010, 2nd edn.) |
| [2] | Mohammad R.,: 'The Electronic Band Structure of III (In, Al, Ga)-V (N, As, Sb) Compounds and Ternary Alloys' (M.Sc. Thesis, Middle East Technical University, 2005) |
| [3] | Hehre W., Random L., Schleyer P., and Pople J.,: 'Ab-initio Molecular Orbital Theory ', (John Wiley and Sons, 1986) |
| [4] | Bouarissa N. and Boucenna M.,: ' Band parameters for AlAs, InAs and their ternary mixed crystals', Phys. Scr., 2009, 79, article id. 015701 |
| [5] | Chimata R.,: 'Optical Properties of Materials Calculated from First Principles Theory', (Department of Information Technology, 2010) |
| [6] | Shimazaki T. and Asai Y.,: 'Energy band structure calculations based on screened Hartree–Fock exchange method: Si, AlP, AlAs, GaP, and GaAs', J. Chem. Phys., 2010, 132, article id. 224105 |
| [7] | Frisch, M.J., Trucks, G.W., Schlegel, H.B., et al.: Gaussian 03, Revision B.01, Gaussian, Inc., Pittsburgh, PA, 2003 |
| [8] | Harker A., Larkins F.,: 'A large unit cell semiempirical molecular orbital approach to the properties of solids. I. General theory', J. Phys. C, 1979, 12, pp. 2487–2495 |
| [9] | M. A. Abdulsattar, 'Self-consistent Field Calculations of Covalent Semiconductors', (Ph.D. Thesis, University of Baghdad, 1998) |
| [10] | Dorsett H. and White A.,: 'Overview of Molecular Modelling and Ab-initio Molecular Orbital Methods Suitable for Use with Energetic Materials', (DSTO Aeronautical and Maritime Research Laboratory, Australia, 2000) |
| [11] | Simons, 'An introduction to theoretical chemistry', (Cambridge University Press, 2003) |
| [12] | Magnasco V.,: 'Methods of Molecular Quantum Mechanics', (University of Genoa, Genoa, Italy, 2009) |
| [13] | F. Jensen, 'Introduction to Computational Chemistry', (2nd Edition, John Wiley & Sons Ltd, 2007) |
| [14] | C. J. Cramer, 'Essentials of Computational Chemistry', (John Wiley & Sons, Ltd., USA, 2004) |
| [15] | Abdulsattar M.A.,: 'Size effects of semiempirical large unit cell method in comparison with nanoclusters properties of diamond-structured covalent semiconductors', Physica E, 2009, 41, pp. 1679–1688 |
| [16] | Demtroder W.,: 'Molecular Physics', (Wiley-Vch , 2005) |
| [17] | Fulde P., 'Solids with Weak and Strong Electron Correlations', (Max-Planck-Institut fur Physik komplexer Systeme, 2008 |
| [18] | Komsa H. and Pasquarello A.,: 'Dangling bond charge transition levels in AlAs, GaAs, and InAs', J. Applied Physics Letters, 2010, 97, article id. 191901 |
| [19] | Mohammad R. and Katircioglu S.,: The Electronic Band Structure of AlN, AlSb, AlAs and their ternary alloys with in', International J. of Modern Physics B, 2006, 20, P. 3199-322 |
| [20] | M. Stadele, M. Moukara, J. A. Majewski and P. Vogl, ' Exact exchange Kohn-Sham formalism applied to semiconductors' (Physical Review B, 59(15), 1999). |
| [21] | S. K. Tewksbury, 'Semiconductor Materials', (Dept. of Electrical and Computer Engineering West Virginia University, 1995). |