-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Condensed Matter Physics
p-ISSN: 2163-1115 e-ISSN: 2163-1123
2012; 2(2): 27-31
doi:10.5923/j.ajcmp.20120202.01
Full-Potential Study of Half-Metallic Ferromagnetism in CdTMO2 (TM=Fe, Co and Ni)
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLS. Amari, S. Méçabih, B. Abbar, B. Bouhafs
Laboratoire de Modélisation et de Simulation en Sciences des Matériaux,Département de Physique Université Djillali Liabès, Sidi Bel-Abbes, Algérie
Correspondence to: S. Méçabih, Laboratoire de Modélisation et de Simulation en Sciences des Matériaux,Département de Physique Université Djillali Liabès, Sidi Bel-Abbes, Algérie.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
In this work, we present an ab initio calculation for the structural, electronic and magnetic properties of CdTMO2 (TM=Fe, Co and Ni) compound. The calculations are performed by a developed full-potential augmented plane wave plus local orbitals (FP-L/APW+lo) method within the spin density functional theory. As exchange-correlation potential we used the generalized gradient approximation GGA form and GGA+U (where U is the Hubbard correlation terms). We have performed total energy calculations of these compounds in the chalcopyrite structure. We present a comparative study between the electronic structures, total and partial densities of states and local moments calculated within GGA and GGA+U. Furthermore, we predict the values of spin-exchange splitting energies Δx(d) and Δx(p-d) and exchange constants N0α and N0β produced by the transition metal 3d states. Our calculations show the half-metallic behavior of these ferromagnetic compounds.
Keywords: DFT, FP- L/APW+Lo, GGA, GGA+U, Magnetic Properties, Spin-Exchange Splitting
Cite this paper: S. Amari, S. Méçabih, B. Abbar, B. Bouhafs, Full-Potential Study of Half-Metallic Ferromagnetism in CdTMO2 (TM=Fe, Co and Ni), American Journal of Condensed Matter Physics, Vol. 2 No. 2, 2012, pp. 27-31. doi: 10.5923/j.ajcmp.20120202.01.
Article Outline
1. Introduction
- Diluted magnetic semiconductors (DMSs) have recently received attention due to their potential usage in magnetic applications including spintronics, magnetic switching, and magnetic recording[1,2]. These materials are alloys of semiconductors in which some cations are substituted by transition metal (TM) ions or rare earth metal ions. The most relevant feature is the coexistence and interaction of two different electronic subsystems: delocalized conduction (s- type) and valence (p-type) band electrons and localized (d or f-type) electrons of magnetic ions. In particular the sp-d exchange interaction leads to strong band splitting, which result in giant magneto-optical effects[3]. The d-d interaction of randomly distributed magnetic ions, triggers formation of spin-glass and anti-ferromagnetic phases, depending on magnetic concentration and temperature[3-6]. Half-metallic DMS are used for injecting spin polarized carrier into semiconductors and spin valves[7]. They show band-gap at the Fermi level for one spin-state[8].In the past decade, a wide range of adamantine semiconducting compounds were known. However, attention was focused on the simplest the III-V and II-VI compounds. Recently increasing attention has been paid to the ternary chalcopyrite compounds because of their potential applications in the field of light-emitting diodes[9], non- linear optic[10], and photovoltaic sensitive material in solar cells[11-13]. The ternary chalcopyrites crystallize in the tetragonal structure type space group I42d (
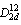 )[14]. Thus the chalcopyrite structure is a superlattice of the zincblende structure (ZnS) by doubling its unit cube along the z-axis that becomes the c-axis of the chalcopyrite structure. The aim of the present study is to investigate the electronic and structural properties of CdTMO2 (TM=Fe, Co and Ni) compounds and understanding the nature and origin of the magnetic interactions. In the following Section, we describe the details of calculations, while in Section 3 we discuss our results. Finally, conclusions will be given in Section 4.
)[14]. Thus the chalcopyrite structure is a superlattice of the zincblende structure (ZnS) by doubling its unit cube along the z-axis that becomes the c-axis of the chalcopyrite structure. The aim of the present study is to investigate the electronic and structural properties of CdTMO2 (TM=Fe, Co and Ni) compounds and understanding the nature and origin of the magnetic interactions. In the following Section, we describe the details of calculations, while in Section 3 we discuss our results. Finally, conclusions will be given in Section 4.2. Computational Method
- The calculations presented in this paper have been performed within the framework of density functional theory (DFT)[15] using the (FP-L/APW+lo) method implemented in the WIEN2K package[16,17]. In this approach space is divided into two regions: non-overlapping muffin-tin (MT) spheres and the interstitial region (IR). Inside the MT spheres, the wave functions are expanded as the superposition of the radial solutions of the one particle Schrodinger equation at fixed energy times the spherical harmonic functions, whereas in the IR the plane wave basis set is used. The plane wave cutoff is determined by RmtKmax = 8.0 (where Rmt is the muffin-tin radius, and Kmax is the maximum modulus for the reciprocal lattice vector). The valence wave functions inside the spheres are expanded up to l= 10. The charge density is Fourier-expanded up to Gmax=14. The self- consistent calculations are considered to be converged only when the calculated total energy of the crystal is converged to less than 1mRyd. The electrons exchange-correlation potential is described in the generalized gradient approximation (GGA) of Perdew et al.[18]. We have considered also the GGA+U scheme (where U is the Hubbard correlation terms). We have used the Coulomb (U) and exchange (J) parameters cited in [19]. We have studied the structural, electronic and magnetic properties of CdTMO2 (TM=Fe, Co and Ni) compounds in the chalcopyrite structure and ferromagnetic phase.
3. Results and Discussion
3.1. Structural Properties
- We have calculated the total energies of CdTMO2 (TM=Fe, Co and Ni) compounds in the chalcopyrite structure within GGA and GGA+U scheme. In order to calculate the ground state properties, we compute the total energies for several lattice constants and fit them with the empirical Murnaghan equation of state[20]. The obtained equilibrium lattice constants, the bulk moduli and their first pressure derivatives of CdTMO2 (TM=Fe, Co and Ni) are given in Table 1. It is observed that the lattice parameters calculated within the GGA+U are larger than those calculated within the GGA. Therefore, the corresponding bulk moduli are smaller.
3.2. Electronic Band Structure and Density of States
- The computed spin-polarized band structures for chalcopyrite CdFeO2 at the calculated equilibrium lattice constant along the high symmetry directions in the first Brillouin zone are shown in Fig. 1. These spectra are calculated for spin-up and spin-down within the GGA including semi-core Cd 4d electrons. The lowest bands derive from the Cd 4d states. Next follow the O 2p derived bands. Between the valence band and the conduction band which consists of Cd-5s states for spin-up and an admixture of Cd-5s and Fe 3d for spin down, there are 3d bands of Fe. The differentiation between majority and minority spins is mainly due to the hybridization of the Fe-3d and O-2p states.The total and partial density of states DOS and PDOS for CdTMO2 (TM=Fe, Co and Ni) obtained using GGA+U are presented in Figs 2-4. For all compounds the d peaks of the TM lie below the Fermi energy EF in spin-up DOS while for spin-down DOS they coincide with the Fermi energy EF. The II–VI materials that contain Fe, Co and Ni are therefore determined to be half-metallic. When the d orbitals of the TM impurity lie in the gap of the host semiconductor, we can clearly see the crystal-field splitting of the d band into the eg and t2g states. However, as we progress through the TM series, the populated d states decrease in energy and hybridize with the host valence band (VB), forming a broad band from which the eg and t2g suborbitals can no longer be clearly distinguished. This behavior is seen in the DOS of all compounds. When we used the GGA+U, we can deduce from theses spectra that the U-Hubbard correlation influences the positions of the electronic states. Fig. 5 depict the difference between the TM d-states calculated using the GGA and GGA+U. We found that the U-Hubbard correlation extensively influenced the TM-d states. One can notice that as the impurity increases in atomic number from Fe to Ni the d peaks of both the conduction and valence bands shift down in energy such that the magnetic ion maintains an integer spin moment.
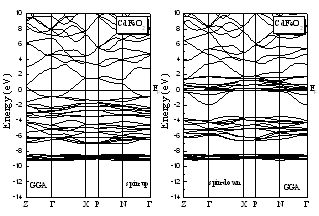 | Figure 1. Spin-polarized band structures for CdFeO2 in the chalcopyrite structure:majority spin and minority spin |
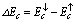 spin-splitting and valence band-edge
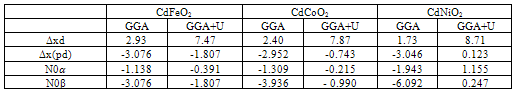
spin-splitting and valence band-edge 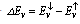 spin-splitting of CdTMO2 (TM=Fe, Ni and Co)using the following relations[21,22]:
spin-splitting of CdTMO2 (TM=Fe, Ni and Co)using the following relations[21,22]: 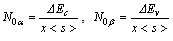 where
where 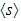 is half of the spin magnetic moment of TM ions and x is TM concentration. The calculated values of N0α and N0β are presented in Table 3. We can deduce the effective d-band exchange splitting Δxd which is defined as the separation between the corresponding spin up and spin down peaks. Another important exchange splitting energy is
is half of the spin magnetic moment of TM ions and x is TM concentration. The calculated values of N0α and N0β are presented in Table 3. We can deduce the effective d-band exchange splitting Δxd which is defined as the separation between the corresponding spin up and spin down peaks. Another important exchange splitting energy is 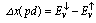 of the top of the valence bands for spin up and spin down. Our results are listed in Table 3. One can notice that the GGA+U values are very large than GGA results. This is due to the fact that the GGA+U extensively influenced the TM-d states.
of the top of the valence bands for spin up and spin down. Our results are listed in Table 3. One can notice that the GGA+U values are very large than GGA results. This is due to the fact that the GGA+U extensively influenced the TM-d states.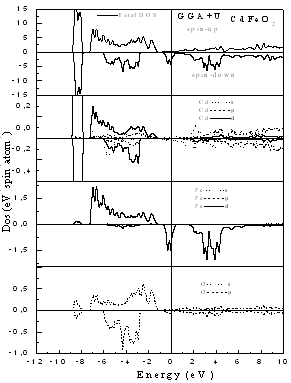 | Figure 2. Spin-dependent total and partial density of states for CdFeO2 using GGA+U |
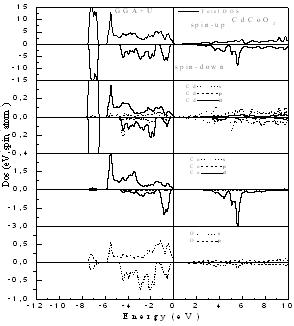 | Figure 3. Spin-dependent total and partial density of states for CdCoO2 using GGA+U |
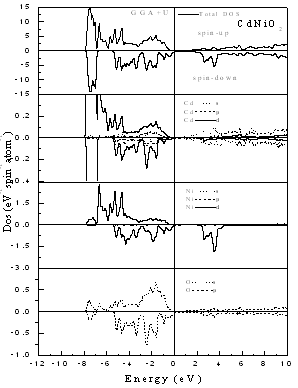 | Figure 4. Spin-dependent total and partial density of states for CdNiO2 using GGA+U |
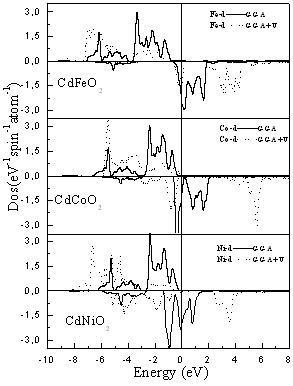 | Figure 5. The difference between the TM d-states calculated using the GGA and GGA+U |
|
 ) within the muffin tin spheres and in the interstitial sites for CdTMO2 (TM = Fe, Co and Ni) using GGA and GGA+U calculations
) within the muffin tin spheres and in the interstitial sites for CdTMO2 (TM = Fe, Co and Ni) using GGA and GGA+U calculations 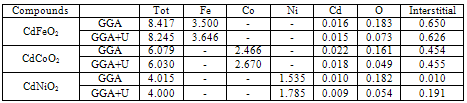
|
3.3. Magnetic Properties
- The calculated total and local magnetic moments for CdTMO2 (TM=Fe, Ni and Co) calculated using GGA and GGA+U are presented in Table 2. The main source of magnetization in these materials is the unfilled transition metal-3d states. The nature of attraction in these diluted magnetic semiconductors can be described by the sp-d exchange splitting. The results show that the total magnetic moments generally come from the TM ions with a small contribution of Cd and O sites. Those magnetic moments are parallel to the TM ions; this may be understood as the tunneling of spin-up impurity states into the neighboring atoms. The p–d hybridization reduces the total magnetic moments of the TM atoms from its free space charge value and produces small local magnetic moments on the non-magnetic Cd and O sites. When we used the GGA+U, we see that the magnetic moments of TM ions increase while those of Cd and O decrease.
4. Conclusions
- In summary, we have performed a first principles calculations based on the FP-L/APW+lo method within the GGA and GGA+U to evaluate the electronic and magnetic properties of CdTMO2 (TM=Fe, Ni and Co). Structural parameters have been computed and it is shown that the lattice parameters calculated within the GGA+U are larger than those calculated within the GGA. The calculated densities of states presented in this study identify the half-metallic behavior of CdTMO2 (TM=Fe, Ni and Co). The U-Hubbard correlation extensively influenced the TM d-states. We found also that GGA+U increases the magnetic moment of TM ions and decreases those of Cd, O and the interstitial sites.The p–d hybridization of TM d (t2g) and O p states reduced the magnetic moments of TM ions from their free space charge values and produced the local magnetic moments on the non-magnetic Cd and O sites. The calculated values of the exchange constants N0α and N0β using GGA+U are larger than those calculated within GGA.
References
| [1] | G. A. Prinz, Science 282, 1660 (1998). |
| [2] | S. A. Wolf, D. D. Awschalom, R. A. Buhrman, J. M. Daughton, S. Von Molnar,M. L. Roukes, A.Y. Chtchelkanova, D. M. Treger, Science 294, 1488 (2001). |
| [3] | Dietl, H. Ohno, F. Matsukura, J. Cibert, D. Ferrand, Science 287, 1019 (2000). |
| [4] | M. Negoita, J. Neamtu, C. V. Onica, OAM – RC 12, 814 (2008). |
| [5] | H. Ohno, F. Matsukura, Solid State Commun. 179, 117 (2001). |
| [6] | Y. Shapira, N. F. O. Jr., P. Becla, T. Q. Vu, Phys. Rev. B 41, 5931 (1990). |
| [7] | G. Y. Gao, K. L. Yao, E. Sasioglu, L. M. Sandratskii, Z. L. Liu, and J. L. Jiang, Phys. Rev. B 75, 174442 (2007). |
| [8] | C. M. Fang, G. A. de Wijs, and R. A. de Groot, J. Appl. Phys. 91, 8340 (2002). |
| [9] | J. L. Shay, L. M. Schiavone, E. Buehier, J. H. Wernick, Appl. Phys. Lett. 43, 2805 (1972). |
| [10] | B. F. Levine, Phys. Rev. B 7, 2600 (1973). |
| [11] | S. Wagner, J. L. Shay, P. Migliorato, H. M. Kasper, Appl. Phys. Lett. 25, 434 (1974). |
| [12] | L. L. Kazmerski, Y. Juang, J. Vac. Sci. Technol. 14, 769 (1977). |
| [13] | B. R. Pamplin, J. Phys. Chem. Solids 25, 675 (1964). |
| [14] | J. L. Shay and J. H. Wernick, Ternary Chalcopyrite Semiconductors Growth, Electronic Properties and Applications (Pergamon Press, Oxford, 1974). |
| [15] | P. Hohenberg, W. Kohn, Phys. Rev. 136, B864 (1964). |
| [16] | P. Blaha, K. Schwarz, G. Madsen, D. Kvasnicka, J. Luitz, WIEN2k code, Technical Universitat Wien, Austria, 1990. |
| [17] | S. Cottenier, Density Functional Theory and the Family of LAPW-Methods: A Step-By-Step Introduction, Instituut voor Kern-en Stralingsfysica, K.U. Leuven, Belgium, 2002. |
| [18] | J. P. Perdew et al. Phys. Rev. Lett. 100, 136406 (2008). |
| [19] | V. I. Anisimov, J. Zaane, O. K. Andersen, Phys. Rev. B 44,943 (1991). |
| [20] | F. D. Murnaghan, Proc. Natl. Acad. Sci. USA 30, 5390 (1944). |
| [21] | C. Echeverria-Arrondo, J. Perez-Conde and A. Ayuela, Phys. Rev. B 82, 205419 (2010). |
| [22] | H. Raebiger, A. Ayuela, and R. M. Nieminen, J. Phys.: Condens. Matter 16, L457 (2004). |