-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Biochemistry
p-ISSN: 2163-3010 e-ISSN: 2163-3029
2021; 11(1): 11-14
doi:10.5923/j.ajb.20211101.02
Received: Sep. 25, 2021; Accepted: Oct. 14, 2021; Published: Oct. 30, 2021

Polymorphism of the Haptoglobin Gene in Homozygous Sickle Cell Patients
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLKandji Pape Matar1, Doupa Dominique2, Djite Moustapha1, 3, Diop Jean Pascal Demba4, Sagne Rene Ngor1, Barry Nene Oumou Kesso1, 3, Thioune Ndeye Mareme1, Mbacke Ndoumbe Mame1, Ndour El Hadji Malick3, Gueye-Tall Fatou3, Seck Moussa5, Diop Saliou4, Lopez-Sall Philomene3, Diop Ndiaye Rokhaya4, Gueye Papa Madieye1, 3
1Laboratory of Biochemistry-Hematology, National University Hospital of Fann, Dakar, Senegal
2Laboratory of Bichemistry, Health Science UFR, Gaston Berger University, Saint-Louis, Senegal
3Laboratory of Pharmaceutical Biochemistry, Faculty of Medicine, Pharmacy, Cheikh Anta Diop University, Dakar, Senegal
4Laboratory of Cytogenetics, Aristide Le Dantec Hospital, Dakar, Senegal
5National Blood Transfusion Center, Dakar, Senegal
Correspondence to: Kandji Pape Matar, Laboratory of Biochemistry-Hematology, National University Hospital of Fann, Dakar, Senegal.
| Email: |  |
Copyright © 2021 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

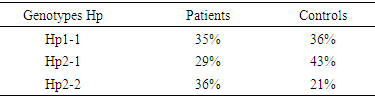
The objective of this study was to determine the frequency of Haptoglobin (Hp) genotypes and their influence on the occurrence of clinico-biological complications of sickle cell disease. This is a prospective case-control study conducted in sickle cell SS subjects. Each patient was matched to a control of the same sex and ± 2 years. Genotyping of the Hp gene was performed by conventional PCR without enzymatic digestion on the Proflex System PCR (Biosystems, Spain) after extraction with QIAmp® genomic kits (Marseille, France). Biochemical parameters were assayed with Cobas c311 (Roche Diagnostics, Switzerland) and NFS ABX Pentra DX Nexus (Machelen, Belgium). The study population consisted of 100 sickle cell SS subjects with an average age of 43 years ± 16 and a sex ratio of 0.75. In sickle cell patients, the frequencies found are respectively of the order of 35%, 29% and 36% for the Hp1-1 Hp2-1 and Hp2-2 genotypes. The prevalence of vaso-occlusive episodes was 55%, with high frequency in patients with genotype Hp2-2. The average hemoglobin level in the controls is 12 g / dl against 8 g / dl in patients (p <0.05). The average C-reactive protein levels were in patients (12.87 mg / l) and controls (5.31 mg / l), the comparison of means showed significant differences. The analysis of the results of the lipid assessment also reveals significant differences. The polymorphism of the haptoglobin gene could be used as a predictor of the severity of clinico-biological complications during sickle cell disease.
Keywords: Sickle cell disease, Polymorphism, Haptoglobin, Lipid metabolism, C-reactive protein
Cite this paper: Kandji Pape Matar, Doupa Dominique, Djite Moustapha, Diop Jean Pascal Demba, Sagne Rene Ngor, Barry Nene Oumou Kesso, Thioune Ndeye Mareme, Mbacke Ndoumbe Mame, Ndour El Hadji Malick, Gueye-Tall Fatou, Seck Moussa, Diop Saliou, Lopez-Sall Philomene, Diop Ndiaye Rokhaya, Gueye Papa Madieye, Polymorphism of the Haptoglobin Gene in Homozygous Sickle Cell Patients, American Journal of Biochemistry, Vol. 11 No. 1, 2021, pp. 11-14. doi: 10.5923/j.ajb.20211101.02.
1. Introduction
- Sickle cell disease is a genetic disorder characterized by the presence of abnormal hemoglobin (HbS), which has the property of polymerizing in its deoxygenated form [10]. This polymerization is the cause of a deformation of sickle red blood cells which is the cause of the physiopathology of the disease. It is the most common hemoglobinopathy in the world; it is widespread especially among black persons [3]. More than 50 million people carry the sickle cell gene; in Senegal the prevalence is 8-10% [15]. Homozygous sickle cell disease (sickle cell SS) is accompanied by an increase in plasma free hemoglobin, which can expose red blood cells to oxidative stress induced by active oxygen derivatives [7]. Thus haptoglobin is the defense mechanism of the body against the deleterious effects of free hemoglobin; it is a protein belonging to the group of α-globin which has the property of setting free hemoglobin [1] in order to protect the tissues against the harmful effects of active oxygen products. However this property of haptoglobin to fix hemoglobin is genotype dependent. Thus, our work aims to study the frequency of the main genotypes of Hp in Sickle cell SS patients and their influence on the occurrence of clinico-biological complications during this pathology.
2. Materials and Methods
- This was a prospective case-control study. The Sickle cell SS patients were recruited from the National Blood Transfusion Center and biological tests were performed at the Biochemistry Laboratory at the Fann National Teaching Hospital Center and Cytogenetics at the Aristide Le Dantec Hospital. For each patient, a control of the same sex and the same age ± 2 years was recruited, non-consenting patients were not included. We studied epidemiological variables (Age, sex), biological (Hemoglobin level, lipid metabolism, C-reactive protein, haptoglobin genotyping) and clinical (vaso-occlusive crisis). Blood samples were taken from subjects fasting, at rest and by venipuncture and collected on an EDTA tube for Hp genotyping. DNA extraction was performed with QIAmp® genomic kits (Marseille, France) and relied on the retention of DNA molecules on a silicate membrane contained in a column. The cells are first lysed with Proteinase K solution and lysis buffer. Then the preparation is passed on a filtration column. Finally, after a washing step which makes it possible to remove contaminants from the sample, the DNA is recovered by elution. Genotyping of the Hp gene was performed using standard PCR with the Proflex System PCR (Biosystems, Spain). The primers A (5'GAGGGGAGCTTGCCTTTCCATTG3') and B (5'GAGATTTTTGAGCCCTGGCTGGT3') are used for the amplification of a 1757pb sequence specific for the Hp1 allele and a 3481pb sequence specific for the Hp2 allele. The primers C (5'CCTGCCTCGTATTAACTGCACCAT3 ') and D (5'CCGAGTGCTCCACATAGCCATGT3') are used to amplify a 349bp sequence specific for the Hp2 allele (KOCH et al., 2002). The PCR was carried out by adding to 2ul of DNA, MgCl2 (1.5mM), 2.5μl of 10mM dNTPs, 12.5µl of Taq polymerase (Applied Biosystems, USA), each of the primers to a volume 1ul and sterile water in a final volume of 25μl. The PCR program used consisted of an initial denaturation at 95°C for 5 min, followed by 35 amplification cycles each comprising DNA denaturation at 95°C for 1 min, primer annealing, and elongation at 69°C for 2 minutes; final elongation was then carried out at 72°C for 10 minutes. The PCR product was visualized on 0.7% agarose gel in the presence of ethidium bromide (EtBr) and a molecular weight marker. The data was collected and used with Microsoft Excel 2013 software. The Student’s t-test was used to compare the averages and the comparison of the proportions was carried out with the Chi2 test and the odds ratio has been used to measure associations. A p-value less than 0.05 was considered statistically significant.
3. Results
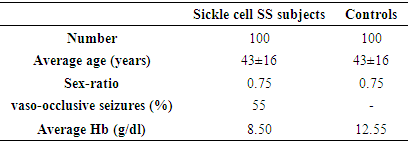
- Our study population consisted of 100 sickle cell SS subjects each matched to a control. The average age of our patients was 43 years old with extreme 15 and 57 years and the sex ratio was 0.75. The prevalence of vaso-occlusive seizures was 55% and baseline Hb was estimated at 8.5g / dl (Table 1).
|
|
|
|
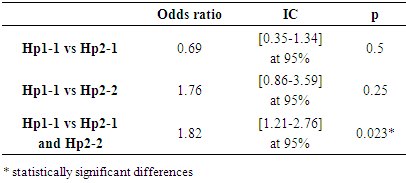
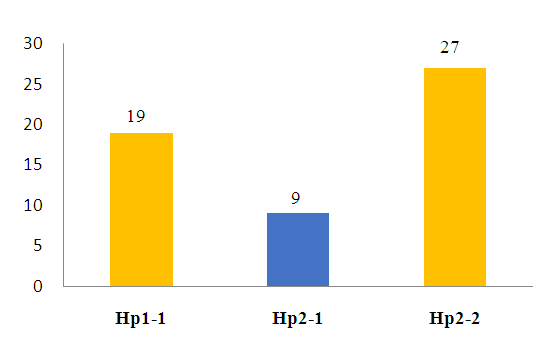 | Figure 1. Prevalence of VOC episodes according to haptoglobin polymorphism |
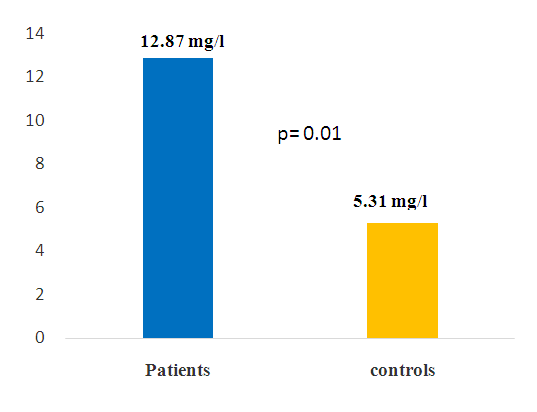 | Figure 2. Mean concentrations of CRP in patients and controls |
4. Discussion
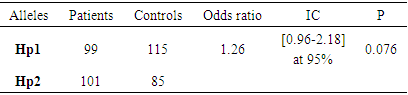
- Sickle cell disease is the most common genetic disease in the black race. The onset of symptoms and complications is associated with several factors that may be environmental or genetic [2-3]. Thus, we set ourselves the research objective of determining the frequency of the different genotypes of haptoglobin in SS homozygous sickle cell patients. The analysis of our results shows an average age of 43, with extremes of 15 and 57 years. This result reinforces the idea that the life expectancy of homozygous sickle cell disease has improved greatly around the world [6]. Jardin et al, in a study conducted in Dakar and involving 40 patients over the age of 15, had returned to an average age of 25.2 years, of which 25% were over 30 years of age [6]. Another study carried out in Senegal by Diop et al shows a result similar to ours [5]. Authors have reported that in the United States, 50% of patients reach the age of 50 years [13] and the life expectancy of patients in Jamaica was estimated at 53 years in men and 58.5 years in women [14]. This increase in life expectancy can be explained by several factors including early diagnosis, the implementation of prophylactic measures, but also the increase of specific centers for the management of sickle cell disease. We found in our study a higher frequency of women with a sex ratio of 0.75. In the literature, we found little significant connection between sickle cell disease and sex. Similar results were obtained by [4]. Genotyping of haptoglobin in controls and sickle cell subjects showed different distributions. The frequency of the Hp2 allele is more increased in sickle cell patients (50.5%) than in control subjects (35.5%). In sickle cell patients, the Hp2-2 genotype predominates with a frequency of 36%. These results confirm those of Gueye et al concerning a study of a series of 68 SS-type sickle cell subjects. They had found a majority frequency of around 36.76% for Hp2-2 [6-7]. It should also be noted that the increased frequency of the Hp2-2 genotype has been found in the literature during cardiovascular diseases but also in the context of other pathologies [9]. The genotyping performed in the control subjects shows frequencies of the order of 43%, 36% and 21% for Hp2-1, Hp1-1, and Hp2-2, respectively. These results are similar to those of Koch et al in a series of 249 subjects. In these subjects, an increased frequency of the Hp2-1 genotype was found with a frequency of 48.2% [11]. In addition, a study in Gambia by Harris et al showed a predominance of the Hp1-1 genotype (30.57%) followed by Hp2-1 (21.66%) and finally 7.01% for the Hp2-2 genotype [8]. In addition, studies have shown a predominance of the Hp1-1 form in Africa; while in Europe the predominant form is Hp2-1 followed by Hp2-2 [12].
5. Conclusions
- These results show an association between homozygous sickle cell disease and Hp2-2 genotype was found at the end of our study. Moreover, studies have shown that this polymorphism could be associated with the severity of complications during sickle cell disease, hence the need to continue this study in order to better understand the impact of these genetic factors on this pathology.CONFLICT OF INTERESTS: The authors have not declared any conflict of interests.CONSENT: Written and informed consent was obtained from each participant.