-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Biochemistry
p-ISSN: 2163-3010 e-ISSN: 2163-3029
2017; 7(5): 83-90
doi:10.5923/j.ajb.20170705.01

Synthesis of 5-[-2(3-Benzyloxy-2-methylamino-propionylamino-3-methyl-pentanoylamino] 6-oxo-heptyl}-carbamic Acid 9H-fluoren-9-ylmethyl Ester (‘VAV-NH-(PEG)23-NH-VAV’, the hydrogel) from NH2(PEG)23NH2 and NCA VAV) and a Study of the Polymerisation Kinetics
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLEstella Judith Salamula1, Misael Silas Nadiye-Tabbiruka2
1College of Natural Sciences, Department of Chemistry, Makerere University, Kampala, Uganda
2Department of Chemistry, University of Botswana, Botswana
Correspondence to: Estella Judith Salamula, College of Natural Sciences, Department of Chemistry, Makerere University, Kampala, Uganda.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Amine terminated Polyethyl glycol (NH2(PEG)23NH2) and 4,8-diisopropyl-6-methyl-1,3,9-oxadiazecane-2,5,7,10-tetrone (NCA VAV) were used to synthesize the hydrogel 5-[-2(3-Benzyloxy-2-methylamino-propionylamino-3-methyl-pentanoylamino] 6-oxo-heptyl}-carbamic acid 9H-fluoren-9-ylmethyl ester (‘VAV-NH-(PEG)23-NH-VAV’). The hydrogel was characterized using proton and carbon NMR together with other spectroscopic techniques. The kinetics of the reaction were investigated in situ in an NMR tube in order to establish the mechanism of the polymerization of 4,8-diisopropyl-6-methyl-1,3,9-oxadiazecane-2,5,7,10-tetrone (NCA VAV with the di-functional diamide poly (ethylene) glycol, as an initiator. The reaction was found to proceed by 1st order mechanism with respect to the NCA VAV concentration.
Keywords: Polyethylene glycol, Hydrogels, N-tert butoxycarbonanydrides-(NCAs’), Polymerization kinetics
Cite this paper: Estella Judith Salamula, Misael Silas Nadiye-Tabbiruka, Synthesis of 5-[-2(3-Benzyloxy-2-methylamino-propionylamino-3-methyl-pentanoylamino] 6-oxo-heptyl}-carbamic Acid 9H-fluoren-9-ylmethyl Ester (‘VAV-NH-(PEG)23-NH-VAV’, the hydrogel) from NH2(PEG)23NH2 and NCA VAV) and a Study of the Polymerisation Kinetics, American Journal of Biochemistry, Vol. 7 No. 5, 2017, pp. 83-90. doi: 10.5923/j.ajb.20170705.01.
Article Outline
1. Introduction
- Hydrogels can be physical, chemical in nature [1] or synthetic. A ‘chemical hydrogel is formed when polyelectrolytes and/or graft polymers of opposite charge precipitate depending on concentration, ionic strength and pH of the solution. Physical hydrogels are formed by 3D hydrophilic polymeric networks and are held together by their molecular entanglements and/ or secondary forces including H-bonding, or hydrophobic forces. These physical bonds, which hold them together, give rise to in-homogeneities as well as chain end defects or chain loops that represent transient network weaknesses in the physical gel. Novel synthetically prepared proteins, like 5-[-2(3-Benzyloxy-2-methylamino-propionylamino-3-methyl-pentanoylamino] 6-oxo-heptyl}-carbamic acid 9H-fluoren-9-ylmethyl ester (Poly VAV-PEGNH-VAV) amide are generally filed into libraries are classified in three groups namely: tryptic, semi-tryptic, and non-tryptic [2]. This classification allows peptides that belong to different tiers to have different “Bonferroni correction factors.”
The scheme improves retrieval performance, significantly, compared to similar correction factors for all qualified peptides, according to their abilities.” [3-5] The wide range of synthetic techniques for peptides allows for the formation of 100-1000’s peptides via an automated sequencer. [6-8] This method has an added advantage over others in that it can be utilized to generate product libraries of high purity in solution and in solid state [9]. However, the main disadvantage is the tedious identifying technique, which necessitates coding and decoding [10]. Other techniques involve a combination of library techniques by mixing beads with peptides [11]. This technique yields reaction volumes in the range of 107-108 peptides for biological and artificial blocks, which is considerably higher than wet synthesis. Condensation polymers include Polypeptides and polyesters, where water or methanol molecules are lost in the synthesis. However, other examples of condensation, such as acrylation polycondensation of π-conjugated C-H bonds in aromatic monomers with dibromo-arylenes are attracting increasing attention as a simple synthetic method in which the preparation of organometallic reagents is not necessary [12].These peptides are then filed into libraries that are split into small peptide mixtures with amino acids at certain spots [13]. The location of defined amino acids and mixed amino acids is known. Therefore all possible combinations are scanned/ screened and all randomized positions are de-convoluted by an interactive process based on library results. [14] Such proteins can then be grafted to Polyethylene glycol as blocks after forming the peptide or synthesized with the polyethylene gycol as an initiator, in much the same way I carried out the reaction.A lot of work has gone into replicating proteins and hydrogels through synthesis using varying methods, forming libraries of these compounds. However, little is known about the kinetics of the reactions. Although the formation of polypeptides through N-tert carboxy anhydrides (NCAs’) is widely known, and is currently used to prepare them, the kinetics and mechanism have received little attention. The only publication that delves into this kinetics study, appears to be that of Waley and Watson. They studied the polymerization of sarcosine N-carbonic anhydride and found the reaction to be complex. The reaction order with respect to (w.r.t) the monomer was initially more than 1 but eventually reached unity towards the end of the reaction, which differed with my NCA-VAV and amine terminated Poly glycol that I used for my experiment. However, when NCA was initiated with preformed polymer, the reaction order, w.r.t to the initiator concentration, was between 1-2 [15].In this article, we report the synthesis and characterization of a chemical hydrogel, which can be used in repair and regeneration in heart tissue, from NCA-VAV and di-functional amine terminated polyethylene glycol (NH2PEGNH2) as initiator. We then investigate the kinetics of this polymerization reaction, utilizing magnetic resonance (NMR) at 600MHz.
2. Discussion
- 5-[-2(3-Benzyloxy-2-methylamino-propionylamino-3-methyl-pentanoylamino] 6-oxo-heptyl}-carbamic acid 9H-fluoren-9-ylmethyl ester (Poly VAV-(PEG)23-VAV amide)1H NMR (600 MHz, DMSOd6): δ 0.89 [14H, t(J=7.4 H), β-(4CH3O)] α-[2CH)], 1.45 [6H,quartet(J=7.4 Hz], β-[2CH3)), 1.72 [1H, q(J=6.6 Hz), α-(2CH), 2.55 [1H, t(J=7.4, 7.2 Hz), CH], 2.67 [1H, t(J=4.0 Hz), αCH], 2.78 [1H, t(J=6.8 Hz), CH], 3.08 [1H, t(J=5.2 Hz), α-CH], 6.10 [1H, s, α-CH], 7.41 [1H, s, NH].In the equation below, the di-functional amine (NH2-(PEG)24-NH2) is used to polymerize the NCA-VAV, to form a hydrogel. Polymerization of NCAs’ proceeds by cationic living mechanism and can be initiated with primary, secondary, tertiary amines and strong bases. [16, 17]. Living polymerisation of NCA VAV to Poly VAV usually proceeds through carbamate and ‘amine’ or activated monomer mechanisms. Carbamate mechanism is initiated by a simple nucleophilic attack on the most acidic carbonyl, and water is released. [18] (See Figure 1 for mechanism and then equation 1).
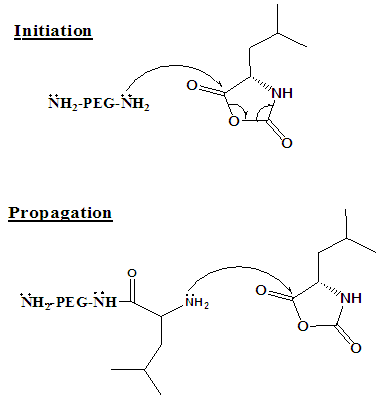 | Figure 1. Illustration of the polymerisation mechanism of a single amino acid NCA |
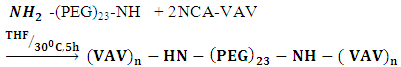 | (1) |
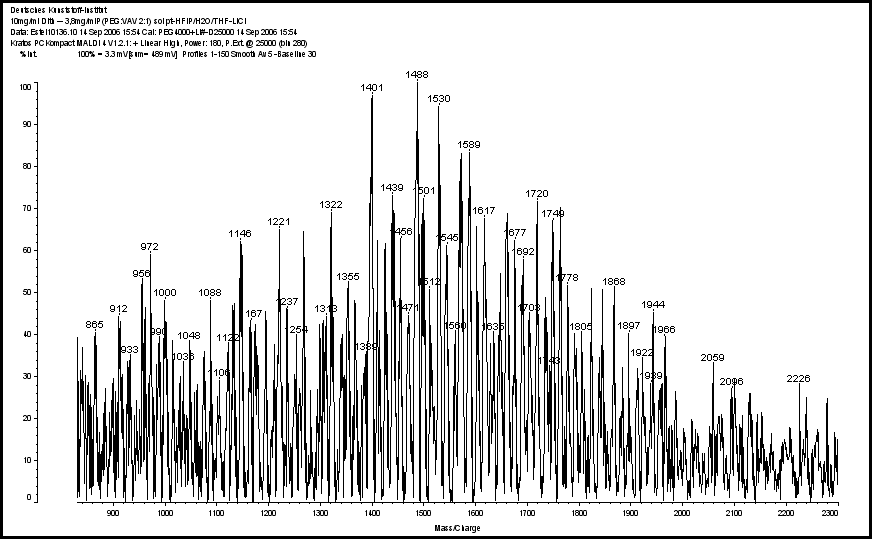 | Figure 2. MALDI spectrum of 2PEG: 1VAV |
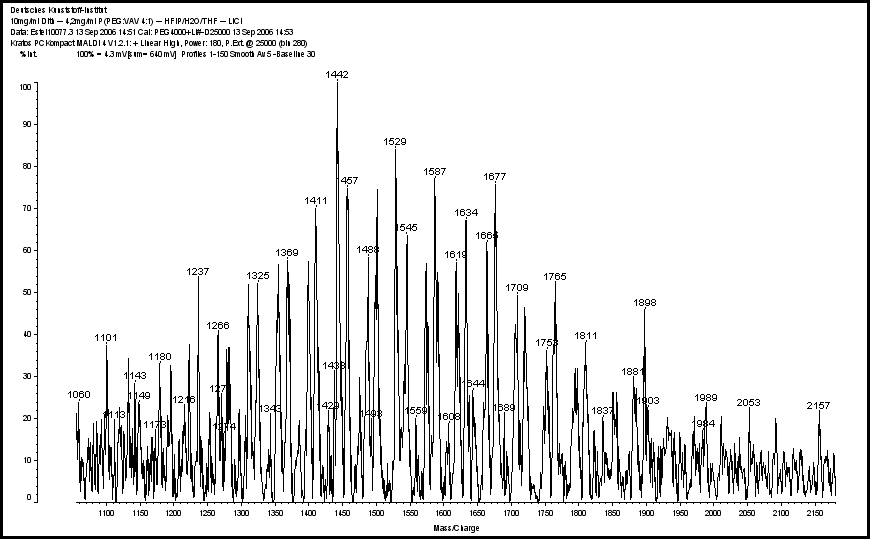 | Figure 3. MALDI spectrum of (4PEG: 1VAV) |
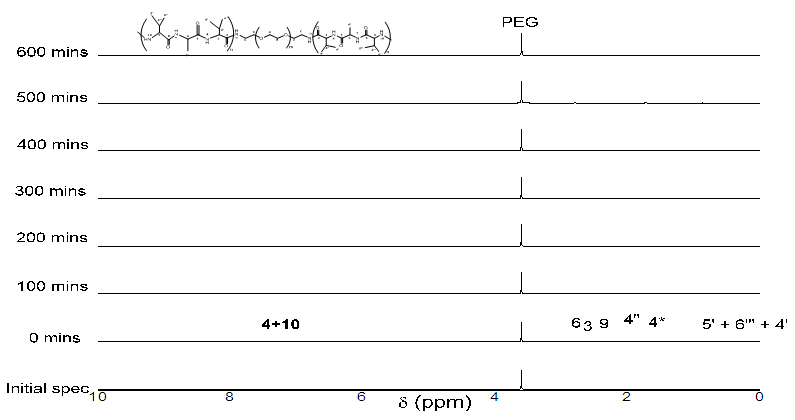 | Figure 4. 1H NMR spectra of reaction between NH2-PEG-NH2 with NCA-VAV against time (mins) (and enhanced spec from 3.0-0.5ppm below) |
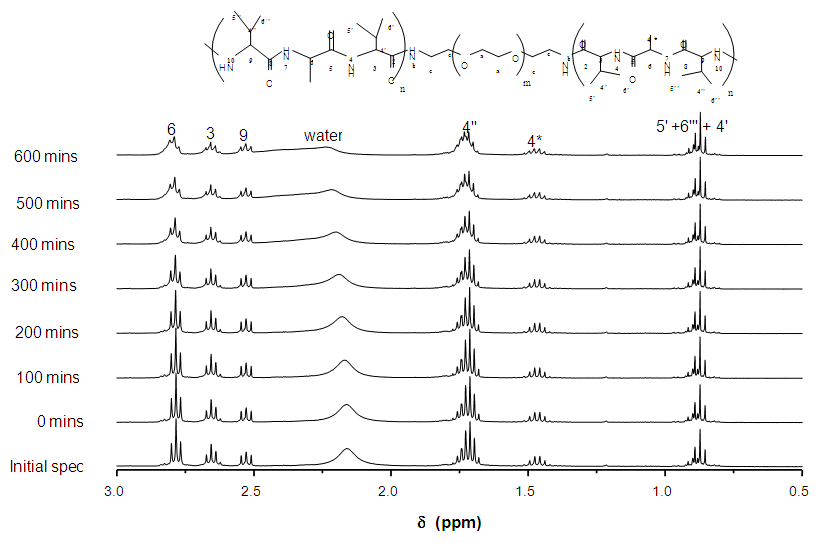 | Figure 5. 1H NMR spectra of reaction between NH2-PEG-NH2 with NCA-VAV over time (mins) and enhanced spec from 3.0-0.5ppm |
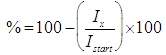 | (2) |
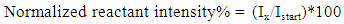 | (3) |
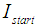 the initial intensity and
the initial intensity and 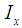 is the intensity, at any given time or concentration. This was taken for designated peaks over the time period. The value was then converted to concentration, since initial and final concentrations of the, 10-diisopropyl-3-methyl-1,4,7-triazecane-2,5,8,9-tetraone (‘NCA-VAV’) were already known. Since the Ix is smaller than Istart the result of equation 2 is a positive % hence we can take the log of the function.Figure 8, shows the change in intensity/concentration ratio Ix/Istart of peaks 4”, 5” and 6” in Figure 5 during the grafting of NCA-VAV onto the H2N (PEG)23 NH2, captured as a function of time, over 10 hours of polymerization. The reaction was initiated as the temperature of the mixture was brought to room temperature (25°C) from 0°C. Propagation and termination (tailing off) occurred when 6, 10-diisopropyl-3-methyl-1,4,7-triazecane-2,5,8,9-tetraone(NCA-VAV) monomer was exhausted. The overall reaction is ‘bimolecular’ probably complex with several steps in the mechanism. Hence, the rates of reaction of the various steps may vary. However, the method was chosen in order to obtain the reaction order, and can be obtained by plotting log (1-(Ix/Istart)*100) against concentration, where a straight line would indicate a first order process at the rate-determining step. The rate of depletion of NCA-VAV as a function of time is obtained by measuring the slope of the tangents of the curve in figure 8 as a function of time or by differentiating the equation of the fitted polynomial with respect to time and substituting in the specific times in the obtained equation, using linear regression equation 4. [19]
is the intensity, at any given time or concentration. This was taken for designated peaks over the time period. The value was then converted to concentration, since initial and final concentrations of the, 10-diisopropyl-3-methyl-1,4,7-triazecane-2,5,8,9-tetraone (‘NCA-VAV’) were already known. Since the Ix is smaller than Istart the result of equation 2 is a positive % hence we can take the log of the function.Figure 8, shows the change in intensity/concentration ratio Ix/Istart of peaks 4”, 5” and 6” in Figure 5 during the grafting of NCA-VAV onto the H2N (PEG)23 NH2, captured as a function of time, over 10 hours of polymerization. The reaction was initiated as the temperature of the mixture was brought to room temperature (25°C) from 0°C. Propagation and termination (tailing off) occurred when 6, 10-diisopropyl-3-methyl-1,4,7-triazecane-2,5,8,9-tetraone(NCA-VAV) monomer was exhausted. The overall reaction is ‘bimolecular’ probably complex with several steps in the mechanism. Hence, the rates of reaction of the various steps may vary. However, the method was chosen in order to obtain the reaction order, and can be obtained by plotting log (1-(Ix/Istart)*100) against concentration, where a straight line would indicate a first order process at the rate-determining step. The rate of depletion of NCA-VAV as a function of time is obtained by measuring the slope of the tangents of the curve in figure 8 as a function of time or by differentiating the equation of the fitted polynomial with respect to time and substituting in the specific times in the obtained equation, using linear regression equation 4. [19]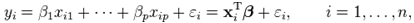 | (4) |
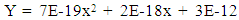 | (5) |
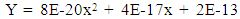 | (6) |
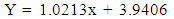 | (7) |
 | Figure 7. Poly (VAV-PEG-VAV) amide, some ten years after it was synthesized |
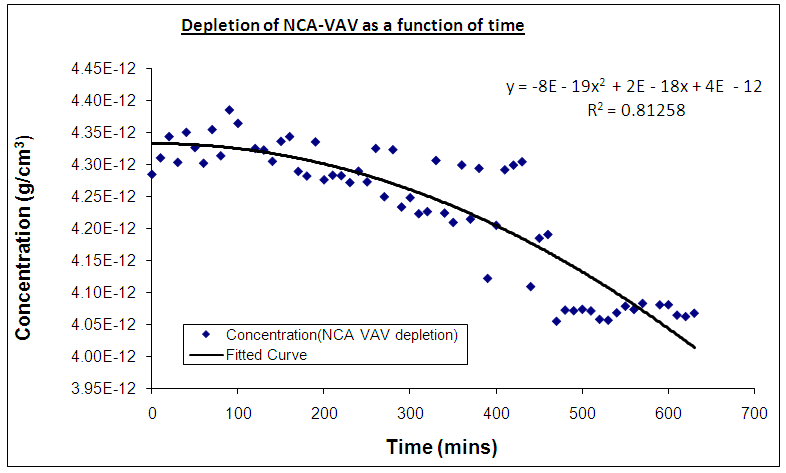 | Figure 8. Depletion of NCA-VAV as a function of time with respect to concentration |
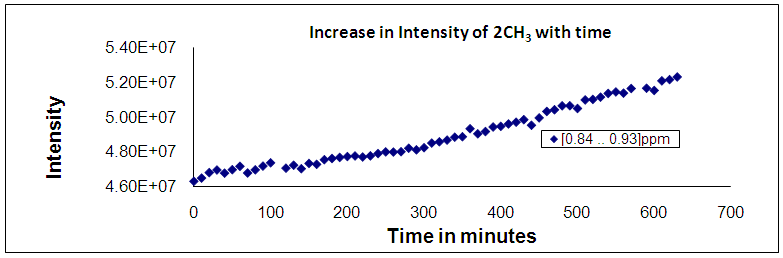 | Figure 9. Shows the variation of product group intensity during grafting polymerization of NCA (VAV). With NH-(VAV)23-NH2 as a function of time with time |
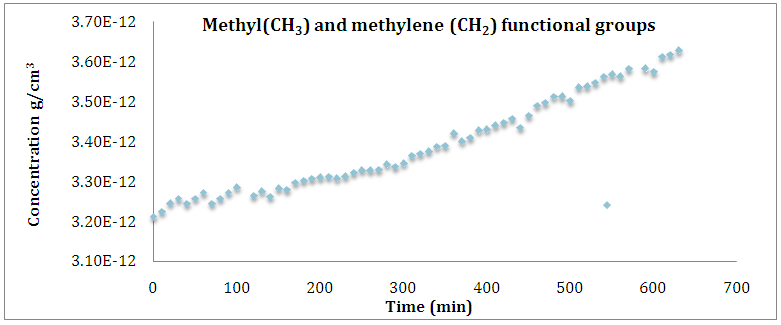 | Figure 10. Illustration of change in product group concentration during grafting polymerization of NCA (VAV). With NH2-(VAV)23-NH as a function of time |
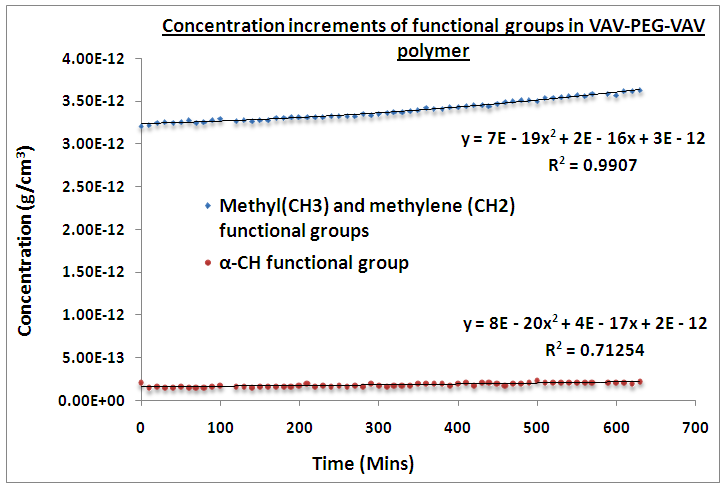 | Figure 11. Study of the rate of reaction of Poly (VAV) amide onto amine terminated PEG as a function of time |
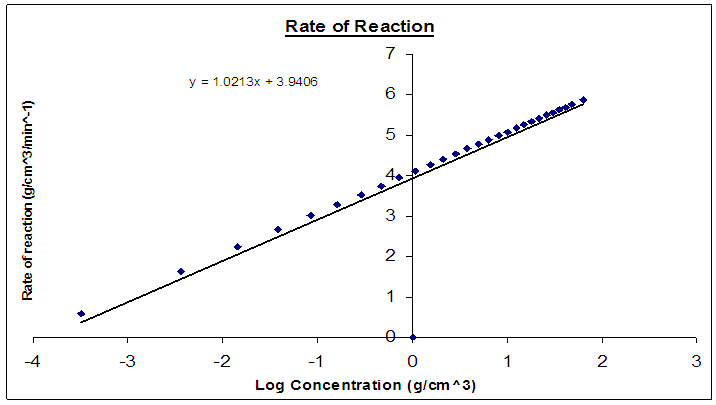 | Figure 12. Rate of reaction of the NCA-VAV depletion as a function of Log concentration |
3. Experimental
3.1. Equipment
- Melting point apparatus (DMA 8000): for melting point determination; TLC paper was used to obtain -Rf values. Nicolet Nexus Fourier-Transform infrared (FTIR): ‘for obtaining fingerprint’ spectrums of molecules. Oxford-NMR (300/400/600MHz Agilent) was used for Carbon and proton Nuclear Magnetic resonance spectra and CapLC-QTOF Ultima ESMS-Electrospray mass spectrometer was used to obtain molecular masses.
3.2. Materials
- Alanine, valine, thionyl chloride, oxalyl chloride, methanol, 4-tert-butoxycarbonyl, litium hydroxide, potassium sulphate, magnesium sulphate, sodium bicarbonate and sodium hydroxide were obtained from Sigma Aldrich; hydroxybenzotriazole (HoBt), silica gel, O-(Benzotiazole-1-yl)-N’N’N’N’-tetramethyluronium tetrafluoroborate (TBTU) were obtained from Advanced ChemTech, South Africa and were used without further purification. THF, DMF, acetonitrile and ethanoic acid, diisopropyl ethylamine were purified by re-distillation before use. Water was obtained from an onsite distilling and de-ionizing system.
3.3. Procedure
- Polymerizing of NCA VAV on NH2-(COCO)23-NH2The experiment was conducted in an NMR tube, in which 0.075 mmol of freeze dried NH2-(PEG)23-NH2 and 0.3 mmol of NCA-VAV were added and allowed to polymerize over a period of 10 hours, in deuterated chloroform. The ‘polypeptide grafting reaction’ was followed by capturing spectral data every 30 min. All changes in intensity were tabulated. Three peaks with significant changes in intensity were studied, tabulated and their intensities plotted as a function of time/concentration. Details of the NMR characterization are listed in the results section.
ACKNOWLEDGMENTS
- The authors wish to acknowledge the contributions from the Department of Chemistry and Polymer science of the University of Stellenbosch in South Africa for the lab space, chemicals and equipment used. The contribution by the laboratory staff is also greatly appreciated.
Abbreviations
- 6,10-diisopropyl-3-methyl-1,4,7-triazecane-2,5,8,9-tetraone/ NCA VAV, Melting point/M.P, Thin layer Chromatography/TLC; Ultra-violet/UV, Infra-red/IR, Nucleic magnetic resonance/NMR; Electrospray mass spec/ESMS.