-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Biochemistry
p-ISSN: 2163-3010 e-ISSN: 2163-3029
2016; 6(2): 37-45
doi:10.5923/j.ajb.20160602.03

Synthesis and Characterisation of 6, 10-diisopropyl-3-methyl-1,4,7-triazecane-2,5,8,9-tetraone/ NCA-VAV
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLEstella Judith Salamula 1, 2, Martin Bredenkamp 3, Misael Silas Nadiye-Tabbiruka 4
1Department of Chemistry, Institute of Polymer Science Stellenbosch University, Stellenbosch, South Africa
2Department of Chemistry, College of Maths and Science, Makerere University, Kampala, Uganda
3Department of Chemistry, Asia International Pacific University, Muak Lek Subdistrict, Saraburi Province, Bangkok, Thailand
4Department of Chemistry, University of Botswana, Gabarone, Botswana
Correspondence to: Misael Silas Nadiye-Tabbiruka , Department of Chemistry, University of Botswana, Gabarone, Botswana.
| Email: |  |
Copyright © 2016 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

NCAVAV= 2-(2-(2-(tert-butoxycarbonyl) methylbutanamido) propanamido)-3-methylbutanoic acid/ Boc-VAV-OH (Trimer) was synthesized by stepwise coupling of different amino acids. The ‘Boc-VAV-OH’ compound was, for the first time, cyclized to give a 6, 10-diisopropyl-3-methyl-1, 4, 7-triazecane-2,5,8,9-tetraone/ NCA-VAV monomer, using a Lewis acid. All compounds were characterized using UV, NMR, melting point, ESMS, TLC and FTIR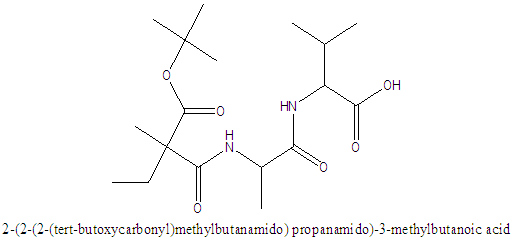
 NCAVAV.
NCAVAV.
Keywords: NCA-VAV, N-tert-butoxycarboanydrides
Cite this paper: Estella Judith Salamula , Martin Bredenkamp , Misael Silas Nadiye-Tabbiruka , Synthesis and Characterisation of 6, 10-diisopropyl-3-methyl-1,4,7-triazecane-2,5,8,9-tetraone/ NCA-VAV, American Journal of Biochemistry, Vol. 6 No. 2, 2016, pp. 37-45. doi: 10.5923/j.ajb.20160602.03.
Article Outline
1. Introduction
- Synthesis of NCAs’ was initially carried out in the 1920s’, using single amino acids and more recently by Deming et al. [1] [2] Thereafter interest in this area increased and the polylactides, polyglycolides and their co-polypeptides such as poly-ε-caprolactone-co-glycolic acid-co-serine (PCGS) [3] and poly-L-lactide-co-glycolide (PLGA)/polyethylene glycol (PEG) [4] were synthesized and are now used as substitutes for proteins. This incorporation of NCAs’ into polyethylene glycol polymers forms the basis for biomaterials [5]. Other applications for these homo- and co-polypeptides include, tissue scaffolding, drug delivery and making of synthetic wool or silk substitutes. [6]More recently, Deming et al [7] synthesized polypeptides using organometallic catalysts, under controlled conditions [8]. With this method, they synthesized polypeptides with molecular weights of up to 25 000 Da with low polydispersities. The group then went on to synthesize block co-polymers including poly-L-lactide-co-glycolide (PGLA) [9] and poly-ε-caprolactone-co-glycolic acid-co-serine (PCGS) [10] using the same catalyst. This was done by, introducing different monomers once the initial monomer was consumed.Currently the three methods used to synthesize “NCA” monomers, are Leuch’s, Fuchs Farthing and ‘Silyazide’. Of these, the most commonly used methods are the Leuch’s and Fuchs Farthing due to the ease of removal of bi-products. The ‘Fuch Farthing’ method utilizes triethyl phosphate, phosgene or its derivatives (triphosgene), at low temperatures, in inert solvents such as dioxane and tetrahydrofuran (THF). [11] Triphosgene (CO(O2CCl3)2 undergoes a nucleophilic attack at the carbonyl carbon, cleaving trichloromethoxy (Cl3CO-) which then dissociates to a chloride anion and a molecule of phosgene, which then reacts immediately [12]. Triphosgene is often used as an alternative to phosgene, as it is safer, easier to handle and to purify [13]. However, triethyl phosphate can be used over shorter time periods and with few side reactions [14-16].Fuchs Farthing is the best method for synthesizing NCAs’ with N-substituted amino acids and amino acids with side groups like histamine (His), ornithine (Orn), lysine (Lys) and threonine (Thr). The reactive functional groups on these amines can be protected with a silane or benzyl (Ar) groups before NCA synthesis, to prevent side reactions that lead to difficult purifying procedures [17].Leuch’s method utilizes Lewis acids to convert the amino acids into acid chlorides e.g., N-ethoxycarbonyl and N-methoxycarbonyl amino acid Chlorides, [18-20] then to NCAs.’ The Lewis acids used include acidic-bromides, such as phosphorus penta-bromide. They cyclize free amino acids more readily than the acid chlorides. [21] (See Scheme 1.1). Dichloromethyl methyl ether and oxalyl chloride were Lewis acids though other reagents such as Dimethylformamide (DMF) and tetrahydrafuran (THF) ether yield pure NCAs. However, in the cyclisation of α-diaminosuccinic acid, oxalyl chloride is preferred, as it does not interfere with the amino acid side groups due to its relatively low reactivity.
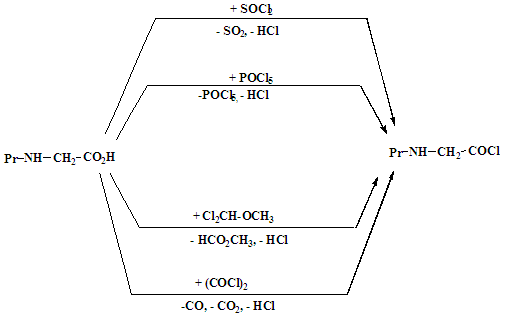 | Scheme 1.1. Synthesis of NCA precursors from various reagents |
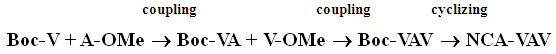 | Scheme 1.2. Synthesis of NCA-VAV/ 6, 10-diisopropyl-3-methyl-1,4,7-triazecane-2,5,8,9-tetraone |
2. Results
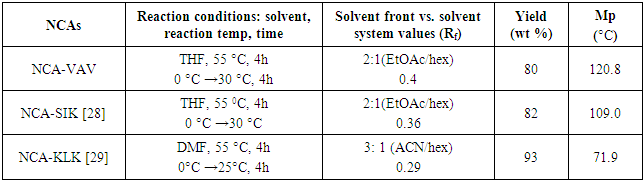
- Synthesis of 2-(2-(2-(tert-butoxycarbonyl)methylbutanamido) propanamido)-3-methylbutanoate/ Boc-VAV-OMeYield: 72%, mp = 73.4°C, Chromatogram-DCM: MeOH, (20:1), Rf = 0.65; Boc-VAV-OMe, MS (EI: 4.15e3 kV) m/ z: 311.19 (2DIPEA+2Na+), 346.2 [14C 24H 5O 5N] +391.3 (M+15H+), 4.2.3 (M+Na+), 416.3 (M+Cl-+5H+); FTIR (KBr discs): 2965 (s/str) CH3, 2876 (s/str) CH2, 2856 (s/str) CH2, 2933 (s/str) CH2, 1464 (m/def) CCH3, 758 (s/ske)C(CH3)2, 1157 (s/ske) C(CH3)2, 1740 (s/str) C=O, 1669 (s/ str) C=O, 1542(s) C=O, 1521(s/str) C=O, 1236 (s/str) CN, 1377 (s/str) CN, 1216(s/str) C-O, 1H NMR (400 MHz, CDCl3): 0.83 (2H, dd(J=3.4, 3.0 Hz), β-(2CH)), 1.07 (2H, dd(J=6.3, 15.8, 6.8 Hz), β-(2CH)), 1.16 (6H, t(J=7.1 Hz), β-CH3), 1.42 (9H, s, α(CH3)3), 2.21 (2H, m, α-(2CH)), 3.71 (3H, s(J=19.0 Hz), α-(O-CH3)), 4.11 (1H, m, α-CH), 4.62 (1H, dt(J=6.5, 14.7 Hz), α-CH), 7.32 (1H, d(J=7.9 Hz), NH), 8.60 (1H, t(J=0.6 Hz) NH), 11.78 (1H, s,NH+), 13C NMR (100 MHz. CDCl3): 16.51 (CH3), 17.8 (CH3), 19.91 (CH3), 19.56 CH3), 29.89 (3(CH3)), 31.10 (CH), 31.17 (CH), 48.89 (CH), 49.1(CH), 52.41 (O-CH3), 57.71 (CH3), 60.19 (CH), 96.72 (C(CH3)3), 157.67 υasy(Boc C=O), 172.11 υsy(Amide C=O), 172.14 (Ala C=O) 173.04 υasy (Ester C=O).The methyl-protecting group on Boc VAV-OMe was removed as described by Joudaiye, J. et al. [24] Synthesis of 2-(2-(2-(tert-butoxycarbonyl)methylbutanamido)propanamido)-3-methylbutanoic acid/Boc-VAV-OHYield: 82%, mp = 155.1°C, Chromatogram-DCM: MeOH (20:1) Rf = 0.22; FTIR (KBr discs): 1496 (m/def) CH2, 1391 (m/def) C(CH3)3, 1366 (m/def), 1418 (m/def) CCH3, 1375 (m/def) CCH3, 1709 (s/str) C=O, 1635 (m/str) C=O, 1506 (m/str) C=O, 1039 (m-w/str) CN, 1164 (m-w/str) CN, 1517 (s/str) COOH; 1H NMR (300 MHz, DMSOd6): 0.74 (12H, m, α-(3CH3). Β-CH3), 1.02 (3H, t(J=7.72 Hz), β-CH3), 1.34 (6H, s, α-(CH3)3, 1.92 (2H, m, α-(2CH)), 2.70 (1H, quartet(J=7.0 Hz), CH), 3.83 (1H, dd(J=6.3, 1.9, 9.9 Hz), CH), 4.01 (1H, dd(J=5.3, 3.1, 5.5 Hz), α-CH), 4.37 (1H, t(J=7.3, 7.2 Hz), OH), 6.61 (1H, d(J=9.1 Hz), NH), 7.73 (1H, d(J=6.3 Hz), NH), 7.90 (1H, d(J=7.4 Hz), NH), 13C NMR (75 MHz. DMSO): 18.76 (CH3), 18.79 (CH3), 19.89 (CH3), 28.09 (C(CH3)3), 30.09 (CH), 31.10 (CH), 46.18 (CH), 48.6 (CH), 58.66 (CH), 78.09 (C(CH3)3, 156.59 υasy (Boc C=O), 172.14 υsy (Amide C=O), 173.14 υasy (Amide C=O), 174.44 υsy ( Val C=O). [25] Cyclisation of 6,10-diisopropyl-3-methyl-1,4,7-triazecane-2,5,8,9-tetraone/ NCA/ VAVYield: 80%, m.p. = 120.8°C, Chromatrogram-EthOAc: hex, (2:1),Rf = 0.4, NCA-VAV MS(EI -1.5e7 kV) m/z: 327.2 (M+H-), 397.2 (M+3Na+), FTIR (NaCl discs): 1435 (m/def) CH3, 2924 (s/str) CH2, 2853 (s/str) CH2, 1587 (s/def) NH, 2490 (w/ str) NH, 2529 (w/str) C(CH3)2, 1174 (s/def) C(CH3)2, 1683 υasy(s/str) C=O, 1698 υsy (s/str) C=O, 1716 υasy (s/str) C=O, 1732 (s/str), 1748 (s/str), 1827 (s/str) O=C-O-C=O, 1790 (s/str) O=C-O-C=O., 1H NMR (150 MHz, DMSO): δ 0.95-1.14 (12H, m, α(3CH3), β-CH3), 1.21 (3H, m, αCH3), 3.61 (1H, m, α-CH), 3.71 (1H, t(J=4.2 Hz), α-CH), 4.25 (1H, q(J=4.8 Hz), β-CH), 8.07 (1H, s, NH), 7.71 (1H, d(J=8.9 Hz), NH), 7.25 (1H, s, NH), 13C NMR (150 MHz, DMSO): 22.6 (CH3), 22.89 (CH3), 24.51 (CH3), 26.10 (CH), 29.20 (CH), 42.00(CH), 45.68 (CH), 51.21 (CH), 169.14 υasy (carbamate C=O), 172.11 (Val C=O), 173.13 υsy (Amide C=O), 174.27 υasy (Amide C=O)Synthesis of 5-(((9H-fluoren-9-yl)methoxy)carbonyl)-2-(2-(6-(((9H-fluoren-9-yl)methoxy)carbonyl) -2-(tert-butoxycarbonyl)hexanamido)-4-methylpentanamido)-5-oxopentanoic acid/ NCA-KLKYield: 93%, mp 71.9°C, ACN/ Hex, (3:1), Rf 0.29: FTIR (NaCl discs): 2940 (s/str) CH3, 1151 (s/ske) C(CH3)2, 1666 (s/str) C=O, 1111 (m-w/ str/str) CN, 1084 (m-w/str) CN, 981 (s/def) Ar, 869 (s/def) Ar, 751(s/ def) Ar, 1H NMR (600 MHz, DMSO): 1.04 [4H, s, β-(2CH2)], 1.83 [2H, s, β-CH2], 2.01 [4H, s, β-(2CH2)], 2.19 [6H, d(J=3.6 Hz), β-(3CH3)], 2.24 [5H, q(J=2.1 Hz), 2CH2, α-CH], 2.40 [2H, s, βCH2), 2.63 (1H, d(J=4.2 Hz), β-CH], 2.79 [1H, s, β-CH], 2.94 [1H, d, β-CH], 3.02 [1H, quartet, β-CH], 3.10 [1H, s, β-CH], 3.61 [1H, d(J=7.06 Hz), β-CH], 3.68 [2H,s, β-CH2], 3.70 [2H, quartet(J=3.4 Hz), CH2], 4.23 [1H, t(J=7.0 Hz), α-CH], 6.06 [1H, s, β-CH2], 7.64 [1H, t(J=0.7 Hz), NH], 7.78 [1H, s, NH], 7.93-8.00 ]16H, s, fmoc], 8.17 [1H, s, NH].
3. Discussion
- Amine functional groups of glycine and valine amino acids were protected with an N-tert butoxycarbonyl group, in basic conditions. The ESMS spectrum of ‘NCA-VAV’ is shown in figure 3, where ‘M’ denotes the ‘compound.' The number of ions bonded to NCA VAV is shown together with the collective charge as salts. FTIR Spectrum of the amine protected amino acids showed deformation vibrations of ‘Boc’ protecting groups C(CH3)3, in the wave-number regions of (1,395-1,38) cm-1. Spectral 1HNMR data for Boc-V-OH have methyl functional groups ‘C (CH3)3' of the ‘Boc group’ which is a characteristically tall peak in the spectrum between wave-numbers 1.3-1.5 ppm for all spectraThe carboxylic functional groups of the alanine and Valine groups were protected with an ester. IR spectrum gave information regarding the presence of twisting, bending and vibrating of the various functional groups in the compounds. The ester groups are found in the wave-number regions of 1,300-1,250 -110 cm-1 and 1,150-1,110 cm-1. After coupling, there was an up-field shift in the amide functional groups to wave-number regions of 1220-1020 cm-1 for the amines, a reliable indication of a successful coupling reaction. Protective esters on the carboxylic acid were not visible at wave-numbers ranging from 1264-1295 cm-1.1H NMR shifts that correspond to the ‘boc’ group for amine and carboxyl protected dimer ‘Boc-VA-OMe’ was found at 1.37 ppm. The amine functional groups of the dimer moved up-field to the 5.0-5.3 ppm range from the 7.5-8.0 ppm range, after the coupling reaction due to the change in the neighborhood.Infrared spectra of trimers have stronger amide peaks owing to the increased number of peptide bonds. The carbonyls, of the Boc ‘C7# protected amino acids have IR peaks in the wave-number regions of 1,395-1,365 cm-1 and methyl groups in the 1,10-1,150 cm-1 and 1,250-1,300 cm-1 region.Important points to note for the Boc VAV-OMe are the ester groups that are discernable in wave-number ranges of 1,700-1,750 cm-1 and 1,150-1,110 cm-1. The esters and amide bonds are masked by the large side groups as the peptide increases in size. This was thought to be the case because the bonds formed when the ester and amide are synthesized are shorter therefore an increase in length of the peptide, through coupling, masks them and makes them less visible in the IR spectrum.1H NMR peaks corresponding to the methyl ester CH3-O, ‘4*’ can be found between 3-4 ppm and NH4+ ‘between 7-12 ppm. The peaks which represent the amide bonds of peptides with methyl esters, ‘4*’ are further downfield (7-11 ppm) in comparison to the amines which are found at (2-3 ppm), owing to the protonation of the amine by hydrochloric acid. The tri-methyl groups of the ‘boc’ protecting groups are found in the range of 1.3-1.5 ppm and the ester group that protects the carboxylic acids in the range of 3.5-3.7 ppm. The methyl groups of Boc-VA-OMe are expressed at 3.7 ppm and for Boc-VAV-OMe at 3.8 ppm. De-protecting BocVAV-OMe gave Boc-VAV-OH which had an elevated melting point and increased values for Rf in comparison to the unprotected trimer. The characterization of the de-protected Boc-VA-OH, using ESMS showed 311.15 (M) +, 431.17 (M + 5Na+ + 6H+), 631.28 (2M)+ sodium salts of Boc VA-OH. A successful de-protection was indicated by the absence of the O-CH3 methyl ‘C4*’ protecting functional group in the IR-spectrum of Boc-GL-OH in the wave-number regions of 1445 and 1460 cm-1.There was little change in positions of the peaks, which represent the ‘Boc’ functional groups of the trimers, which are typically found in the ranges of 1.3-1.4 ppm. The characteristic peak for methyl-protecting group which is typically found in the range of 1445(s) and 1460(s) for Boc VAV-OMe, respectively, is absent after the de-esterification.Yields of the de-esterified dimers and trimers were greater than 60% with melting points that were generally higher than protected trimers. IR spectral data of de-protected trimer.‘Boc VAV-OH,’ shows were successful removal of the methyl-protecting group ‘C4*’ from the carboxylic acid, had occurred. The methyl-protecting group, which is typically refected in the wave-number regions of 1310-1250 cm-1, is absent. 1HNMR experiments were run on the 400 and 600 MHz NMR instrument, using deuterated chloroform as solvent. The biggest difference between the spectral data of the protected trimers is the lack of the ‘C4*/ ‘CH3 - O’ methyl functional group, which protects the carboxylic acid.Melting points of N-tert-butoxycarbonylanhydrides (NCAs’) are generally lower than melting points of their precursors, possibly due to the packing of the rings. The ESMS spectra for the NCAs' were taken at a very low negative voltage to avoid splitting the rings. The spectrum comprises of various ions and salts of the structures and is shown in Figure 3. ‘M’ denotes the NCA. Chloride and sodium ions form salts with the NCA and their charges are indicated by their chemical symbols, below the spectrum in the subject line.The IR spectrum of the NCA shows the fingerprints of the molecular structure and functional groups with slight shifts up-field, for the carbonyl and carbamate of wave-number ranges of 1,710-1,690 cm-1, with strong CO-NH signals in 1220-1020 cm-1 range. There was an absence or reduced signals for the amino acid in the wave-number regions of 3500 cm-1, an absence of the ‘Boc’ protecting group, in the wave-number region of 1395-1385 cm-1 and the presence of the anhydrides in the wave-number region of 1840-1740 cm-1, in figure 1.
 | Figure 1. FT-IR NCA-VAV/ 6, 10-diisopropyl-3-methyl-1,4,7-triazecane-2,5,8,9 tetraone |
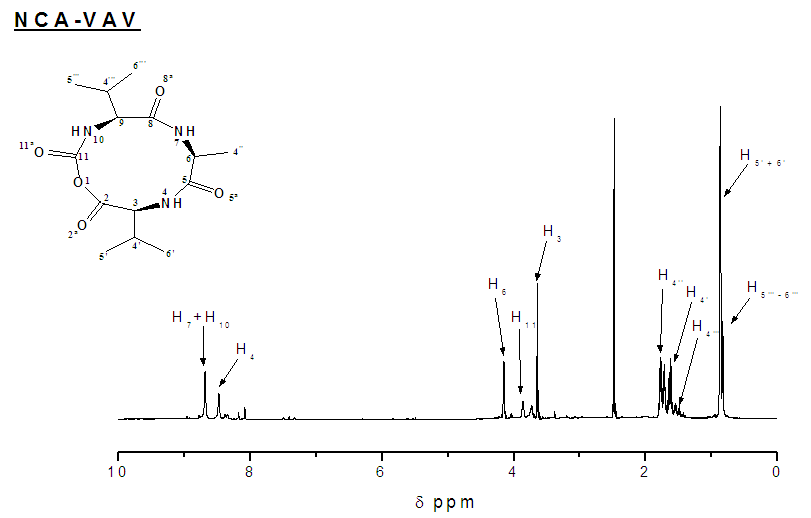 | Figure 2.  of NCA VAV/ 6, 10-diisopropyl-3-methyl-1,4,7-triazecane-2, 5, 8, 9-tetraone of NCA VAV/ 6, 10-diisopropyl-3-methyl-1,4,7-triazecane-2, 5, 8, 9-tetraone |
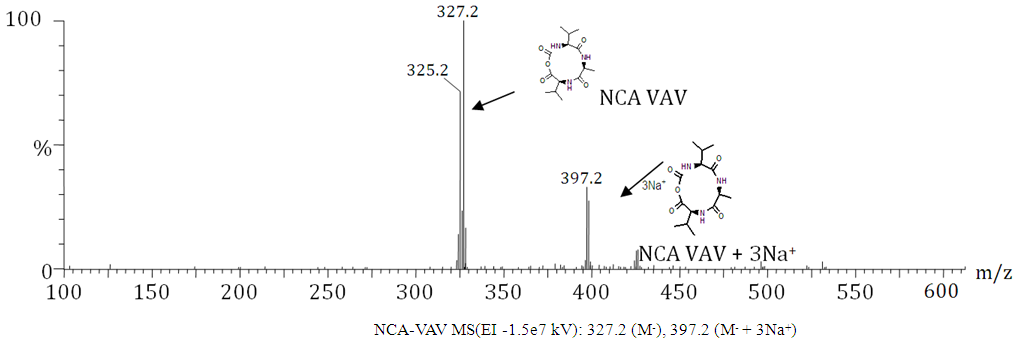 | Figure 3. ESMS of NCA-VAV and its salts |
|
4. Experimental
4.1. Equipment
- Perkin Elmer melting point apparatus (DMA8000), nexus FTIR, Oxford NMR model (Agilent), CapLC coupled to Q-TOF Ultima ESMS.All L-amino acids and peptides were characterized by their melting points and Rf values (product front vs. solvent front), and by nexus FTIR, Oxford-NMR (300-600 MHz) and Proteomics using a CapLC coupled with a Q-TOF Ultima ESMS. The melting points were observed with Perkin Elmer, melting point instrument (DMA 8000). Product front vs. solvent front was obtained from thin layer chromatography (TLC) using appropriate solvent systems. Nicolet Nexus Fourier-Transform infrared (FTIR) was used to characterize ‘fingerprint’ spectra of organic molecules. A nexus FTIR instrument was used under inert conditions to improve the signal: noise ratios, and obtain better spectra. All the above instruments are at The University of Stellenbosch, South Africa. Molecular masses were obtained using time-of-flight electro-spray mass spectrometry (TOF-ESMS). [30, 31] Ionization by spray techniques are based on desorption of the ions of a compound from a chromatographically separated liquid solution, to a mobile phase that contains an electrolyte.
4.2. Materials
- Alanine, valine, thionyl chloride, oxalyl chloride, methanol, 4-tert-butoxycarbonyl, litium hydroxide, potassium sulphate, magnesium sulphate, sodium bicarbonate and sodium hydroxide were obtained from Sigma Aldrich; hydroxybenzotriazole (HoBt), silica gel, O-(Benzotiazole-1-yl)-N’N’N’N’-tetramethyluronium tetrafluoroborate (TBTU) were obtained from Advanced ChemTech, South Africa and were used without further purification. THF, DMF, acetonitrile and ethanoic acid, diisopropyl ethylamine were purified by re-distillation before use. Water was obtained from an on site distilling and de-ionizing system.
4.3. Procedure
- The amine functional group of the first L-Valine (1A) was protected with 4-tert-butoxycarbonyl (boc) [32] [33] to form a Boc-Val (1B). [34] The crude product was purified by chromatography over silica gel (0.06nm, DCM: MeOH), Rf = 0.28, and gave white crystals of m.p. 97°C. [35] (Yield of 62%, see Figure 2). The other amines (2A and 4A) L-Valine and L-Alanine were protected with methoxy groups in a solution of thionyl chloride and methanol, at 0°C. [36] The reaction afforded clear white solid crystals of methyl 2-aminopropanoate/ A-OMe, (m.p of 108°C, yields of 66%). HPLC showed purity of 99%; TLC Chromatogram-DCM: MeOH, 20:1, Rf = 0.2. Methyl 2-amino-3-methylbutanoate/ H-V-OMe gave yields of: 94%, mp = 98.8°C, TLC Chromatogram-DCM: MeOH (20:1) Rf = 0.65.The, 2B gave yields = 81.1%, M.P. = 108°C, Chromatogram: 20DCM: 1MeOH, Rf=0.2 [37], 4B [38], which gave a yield = 94%, M.P = 98.8°C, Chromatogram: 20DCM: 1MeOH, Rf = 0.65. The protected amines were then coupled with TBTU [39] in an alkaline solution, to form protected dimer (3A) and gave yield = 94%, M.P. = 98.8°C, Chromatogram: 20DCM:1MeOH, Rf = 0.75. [40] and trimers (5A), as described in scheme1.5. [42]
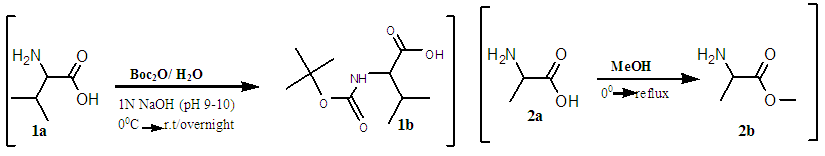 | Scheme 1.3. Protecting amine groups on amino acids using a tert-butoxycarbonyl for the amine and methoxy for the carbonyl |
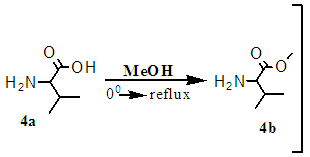 | Scheme 1.4. Protecting carboxylic functional group with methoxy group |
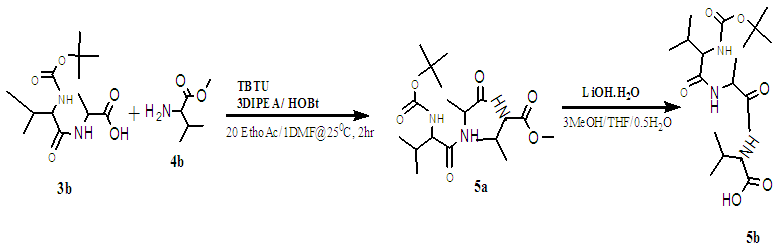 | Scheme 1.5. Coupling of partially protected dimer Boc Val-Ala-OH and Val-OMe then de-protected to give Boc Val-Ala-Val-OH. [42] |
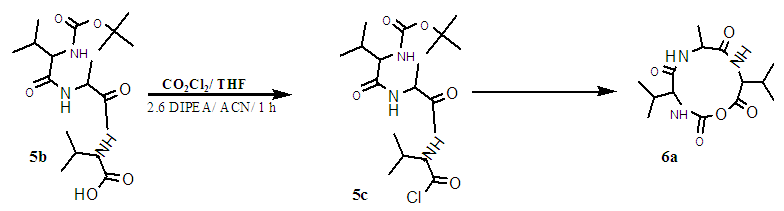 | Scheme 1.6. Cyclisation of timer to NCA VAV |
ACKNOWLEDGEMENTS
- The authors wish to thank the staff at the University of Stellenbosch, particularly, Mrs. E. Cooper and Mrs. M. Hundall, for their contribution to this work. They would also like to thank the National Research Foundation of South Africa for funding the research, Professor Harald Pasch and colleagues at the Deutsches Kunkstoff-Institut for all the analysis and funding of my work in Germany. Finally the contributions made by the department of Chemistry and Polymer Science and the University of Stellenbosch library, are gratefully acknowledged. Last but not least, we wish to register our gratitude and acknowledgement to the project promoter the late Ronald. D. Sanderson for his encouragement and contributions to the work; may his soul rest in eternal peace.
Abbreviations
- 2-(2-(2-(tert-butoxycarbonyl)methylbutanamido) propanamido)-3-methylbutanoate = Boc-VAV-OMe, 2-(2-(2-(tert-butoxycarbonyl)methylbutanamido) propanamido)-3-methylbutanoic acid = Boc-VAV-OH; 6, 10-diisopropyl-3-methyl-1,4,7-triazecane-2,5,8,9-tetraone = NCA VAV