-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Advances in Computing
p-ISSN: 2163-2944 e-ISSN: 2163-2979
2012; 2(2): 23-28
doi: 10.5923/j.ac.20120202.04
Comparison of Structure Based Sequence Alignment Programs for Protein Domain Superfamilies with Multiple Members
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLA. Gandhimathi , Anu G. Nair , R. Sowdhamini
National Centre for Biological Sciences (TIFR), UAS-GKVK Campus, Bellary Road, Bangalore 560065, India
Correspondence to: R. Sowdhamini , National Centre for Biological Sciences (TIFR), UAS-GKVK Campus, Bellary Road, Bangalore 560065, India.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Structure comparison is used to reveal the similarity between protein structures. Every method has its own strength and weakness and the assessment parameters need to be appropriate to the original question on performance of the method. Here, we have assessed three multiple structure-based sequence alignment programs and compared their results. The results suggest that superfamily members which have low sequence identity (<40%) can be aligned using flexible structure alignment methods followed by methods which consider multiple structural features like COMPARER. This kind of structural analysis protocol appears to produce more relevant results, due to consideration of large number of structural features, rather than pure geometric features.
Keywords: Structure Alignment, Outliers, Domain swapping, protein evolution, distant relationships
Article Outline
1. Introduction
- Protein sequence alignments are important in understanding the structural, evolutionary and functional relationship between proteins[1]. It will be more challenging to perform alignments in distantly related proteins owing to high sequence divergence. Computation of structure-based alignment is a delicate task, but can give rise to more reliable alignments at distant relationships, when compared to pure sequence alignment[2].Any method for protein structure alignment needs to balance coverage versus accuracy. Some methods align the core of a protein at very high accuracy (i.e., very low RMSD) and very low coverage (i.e., omitting loop regions), while some methods prefer to increase the coverage (i.e., include the loop regions in the alignment) by compromising on the accuracy (i.e., increasing the RMSD) [3-7]. Structure alignment programs are preferred since they have high accuracy, high coverage and fast execution to cope with the increasing number of structures. Certainly, maximizing the biological relevance of a result is going to be the most desirable outcome in a majority of the cases.In general, there are a number of structure alignment programs available. Some structure alignment programs are developed for aligning a pair of 3D structures called as pairwise structure alignment programs (PStA) and some programs are used for multiple structure alignment (MStA). Examples for PStA methods are LSQMAN[8], FATCAT[9], MINRMS[10], and Dali[11]. MStA Programs are MASS[12], Matt[13], MultiProt[14], MUSTANG[15], POSA[16], SALIGN[17] and 3DCOMP[18]. COMPARER[19] is another structure-based sequence alignment program which considers various structural features to recognize the structural core and variable regions to guide the presence of gaps and to obtain reliable alignments. But the program needs initial equivalencies from any of the alignment program or through graphical inspection. Any sequence or structure alignment can be refined through COMPARER to obtain final alignment.In a previous study, we investigated alignment accuracy of several frequently used structure based sequence alignment methods[20].Three thousand and fifty-two alignments of 218 pairs of protein domain structural entries, with <40% sequence identity, belonging to different structural classes, of diverse domain sizes and length-rigid/variable domains were performed using 12 programs. These programs were compared and assessed by three structural parameters such as root mean square deviation, secondary-structural content and equivalences. The biological and functional relevance of such alignments were examined for some examples. From this study, we concluded that FATCAT, MATT, DALI, MINRMS and LSQMAN programs perform equally in most of the cases. In many cases, LSQMAN fails to improve the percentage of secondary-structure equivalences. This study helped us to select the suitable tool MINRMS for aligning two member superfamilies, where only two domains exist in the superfamily with <40% identity[21].
2. Analysis of Multiple Structure Alignment Tools
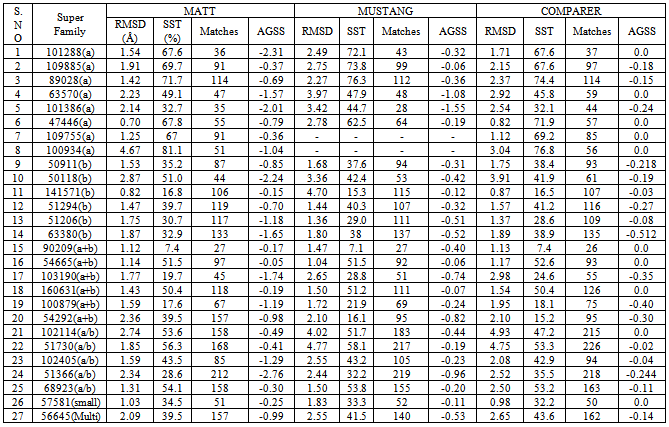
- In case of multiple structure alignment, a program that can produce high-quality alignment, flexibly handle multi-chain structures, process a large number of input structures and provide a consistent output format (like exactly reference to PDB residue numbers) is desirable. For a broader applicability, proper treatment of local conformational variability is probably of utmost importance. We have originally started with the comparison of six different methods for the alignment of multiple members. Our main interest was to understand the structural and functional relationship of superfamilies where the members have <40 identity. Hence, we would prefer structure-based sequence alignment and consider the whole domain for alignment. Based on our criteria, only two programs are suitable for multiple structure alignment (Table.1). We have assessed the two programs to choose the best program to align aforementioned task.
3. Features of Structure Alignment Programs
- It is always a challenging task to compare all the existing programs since they have limitations at various levels. Hence it is better to compare the programs which are suitable for particular type of data. In our analysis, we found MATT, MUSTANG and COMPARER programs are suitable (Table 1) by highlighting the limitations of other programs for aligning domains at superfamily level. MUSTANG aligns residues on the basis of similarity in patterns of both residue–residue contacts and local structural topology. MATT allows local flexibility between fragments: small translations and rotations are temporarily allowed to bring sets of aligned fragments closer, even if they are physically impossible under rigid body transformations.Current methods for structure comparison and alignment usually focus on optimizing geometrical similarities between two or more structures. The alignment of superfamily members are based on the conservation of structural features such as secondary structures, hydrogen bonding and solvent accessibility. Hence, apart from geometrical features, parameters that reflect secondary, tertiary, possibly quaternary features and evolutional information can help in finding the most relevant alignment between structures. Thus, COMPARER is one such method that takes account of all the above mentioned parameters.
4. Assessment Parameters
- In the earlier study[20], we had employed three methods such as RMSD, POSSE (Percentage of Secondary Structure Equivalences) and number of fitted points for assessing the alignment quality and accuracy. These parameters are well explained by our earlier work. We have used one more parameter AGSS (Assessment of gaps in secondary structure) which again depends on the structural information representing the number of gaps introduced within secondary structures during the alignment. If there are more gaps within secondary structure blocks, then the quality of the alignment is questionable.AGSS describes the number of gaps which are introduced within secondary structures in the course of alignment.AGSS= -[(G1+ G2+ …… + Gn)/n]where,n = number of regular secondary structures in the alignment, Gk = (no. of gaps in K)/(length of K, where K is the kth secondary structure.
5. Structure Based Sequence Alignment
- Initially, superfamily members are aligned by MATT, MUSTANG and the alignment is annotated by JOY program[22]. Since COMPARER needs initial equivalences and guided tree for the alignment, we primarily compared MATT and MUSTANG for initial alignment. There are total of 731 superfamilies are aligned using both the methods (data not shown). MATT can handle all kinds of superfamily like total number of domains (2-239) and size of the domain. But, MUSTANG is not suitable for handling multi-chain proteins. Around 86 superfamilies have high RMSD between superimposed structures and failed to align large superfamilies due to some technical issues. For performing refined alignment using COMPARER, initial equivalences are taken from both MUSTANG and MATT. After the COMPARER alignment, MNYFIT[23] is used to obtain superimposed structures. There are 25 superfamilies, belonging to different classes, were used for assessing the accuracy of structure comparison and alignment by MATT, MUSTANG and COMPARER. The alignments were checked using the assessment parameters as mentioned above.
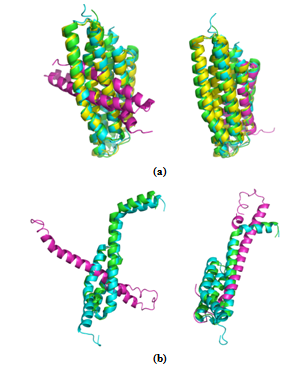 | Figure 1. MUSTANG and MATT comparison. Protein domains that belong to a superfamily are shown in the best fit form after superposition.(a) Superfamily Phou-like (SCOP code: 109755) (b) Superfamily Heat shock protein 70kD (HSP70), C-terminal subdomain (SCOP code: 100934). Result of superposition obtained by MUSTANG is shown to the left and that obtained by MATT is shown to the right. In both these examples, results obtained by MATT are better |
5.1. Predicting Structurally Deviant Members
- The suggested protocol provides good alignment accuracy with low RMSD. It still permits us to find structurally deviant members of the superfamily which are called as outliers. Glutathione synthetase ATP-binding domain-like superfamily (SCOP code: 56059) contains 22 domains with low sequence identity (<40%). Alignment protocol using MATT followed by COMPARER yielded satisfactory structure-based sequence alignment of these superfamilies, leaving only two members (d1eucb2,d2nu7b2) with high RMSD (>5.5Å). These two members belong to Succinyl-CoA synthetase, beta-chain, N-terminal domain family (Figure 3). The next member (d1kbla3) with high RMSD belongs to pyruvate phosphate dikinase family. It is already reported that two families (succinyl-CoA synthetase and pyruvate phosphate dikinase) from the glutathione synthetase ATP-binding domain-like superfamily are slightly more different than the other families. These two families display the same manner of binding the nucleotide inside the active site[24]. In these cases, structural alignment of these outlier members was still possible.
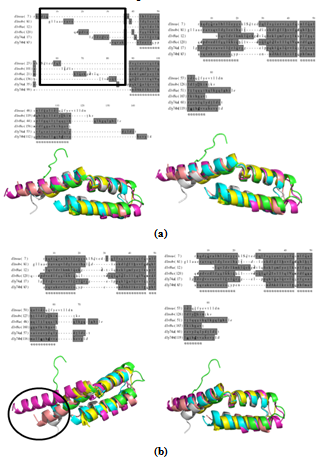 | Figure 2. shows the structure alignment of L27 domain (SCOP ID 101288). Fig 2(a) shows MATT derived alignment and the structural superposition (left) and COMPARER refined alignment and the structural superposition (right). More number of gaps are introduced in the MATT alignment shown in highlighted box. Fig 2(b) shows the same as Fig. 2(a) but for MUSTANG-derived alignment. Alignment is improved, as shown in highlighted circle, with less number of gaps |
|
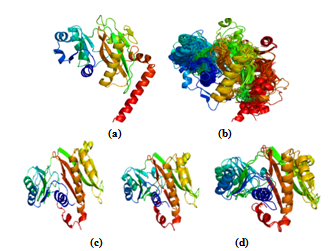 | Figure 3. Identifying structurally deviant members in Glutathione synthetase ATP-binding domain-like superfamily. (a) & (b) shows the representative structure of this superfamily and alignment of 20 protein domains. (c) &(d) shows the two outliers belonging to Succinyl-CoA synthetase family |
5.2. Domain Swapping –TorD like Superfamily
- Domain swapping is an important and interesting phenomenon which refers to two or more proteins exchanging equivalent parts of their structures to form intertwined oligomers, inclusive of dimers[25]. Sequence-based alignment tools may be inadequately sensitive to detect such evolutionarily distantly related members. Moreover, conventional structure comparison algorithms are weak to detect global similarities between proteins related by domain swapping due to considerable difference in the structure of swapped and non swapped forms[5,26].TorD like superfamily in PASS2 dataset has four members, of which three are having closed monomer and one member (d1n1c_) is in open domain swapped conformation[27]. The 3D structures show a long loop region separating the N- and C-terminal domains of the proteins. This loop retains the highly conserved ‘E(Q)PxDH’ motif[28] as equivalent when aligning a ‘‘closed’’ monomer and its domain-swapped ‘‘open’’ homologue. Many structure alignment programs tend to yield an alignment of monomers with only one domain of the swapped dimer aligned. MATT is one such program which gives alignment of monomers with one domain, but COMPARER refined alignment retains both the swapped as well as non-swapped domains as equivalent. Such biologically meaningful alignment proves to be helpful to understand the relationship between monomers and swapped dimers. MUSTANG takes care of domain swapping problem in the alignment. Apart from this, MUSTANG and COMPARER refined alignment retains the linker motif in the alignment as equivalent regions both in the monomer and domain-swapped form despite their structural differences (Figure 4).
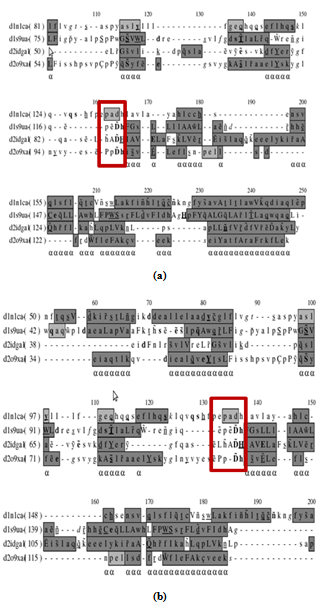 | Figure 4. (a) shows the alignment of TorD superfamily by MUSTANG. MUSTANG is able to align the linker conserved motif (E(Q)PxDH) of swapped and non-swapped domains. Figure 4(b) shows COMPARER- refined alignment where initial equivalences are from MATT. MATT fails to align the conserved motif although MATT followed by COMPARER gives rise to an alignment where the conserved motif is equivalent |
6. Conclusions
- Structure-based sequence alignment methods are crucial for understanding distantly related proteins. We have mainly compared structure comparison tools which give rise to consistent and reliable alignment of multiple members that in turn can enable better understanding of the structural, evolutionary, functional relationships between distantly related proteins at the superfamily level. The aim of this study is not to rank or benchmark the different methods, but instead to recognise the differences in the results and the challenges that remain.We found that an alignment protocol with MATT followed by COMPARER works well for most of the superfamilies. Although MUSTANG performed equally well in many cases and can handle alignment of swapped domain with monomers, it failed to align two superfamilies in our test dataset of 25 superfamilies that belong to alpha-class. MATT could handle all 25 superfamilies, but it introduces lot of gaps in the alignment. Simple RMSD measures are insufficient to recognize good quality alignments. Structure comparison programs like MATT face challenges in swapped domain examples, unlike MUSTANG. However, in all superfamilies studied, COMPARER is efficient in improving the alignment and in recognising the highly deviant members, namely the outliers, of the superfamily.
ACKNOWLEDGEMENTS
- A.G is supported by Senior Research Fellowship from the Council of Scientific and Industrial Research (CSIR) Government of India.
References
| [1] | Sierk ML, Kleywegt GJ, “Deja vu all over again: finding and analyzing protein structure similarities”, Structure, vol.12, no.12, pp.2103-2111, 2004. |
| [2] | Carugo O, “Recent progress in measuring structural similarity between proteins”, Current Protein & Peptide Science, vol.8, no.3, pp.219–241, 2007. |
| [3] | Godzik A, “The structural alignment between two proteins: is there a unique answer?” Protein Science, vol.5, no.7, pp.1325 –1338, 1996. |
| [4] | Christoph B, Christine S. S, Peter L, “Accuracy analysis of multiple structure alignments”, Protein Science, Vol.18, PP.2027-2035, 2009. |
| [5] | Liu W, Srivastava A, Zhang J, “A Mathematical Framework for Protein Structure Comparison.” PLoS Computational Biology, vol. 7, no.2, e1001075, 2011. |
| [6] | Aysam G, Knapp E, “GIS: a comprehensive source for protein structure similarities”, Nucleic Acids Research, Vol. 38, pp. W46–W52, 2010. |
| [7] | Joseph A.P, Srinivasan N, Alexandre G, “Improvement of protein structure comparison using a structural alphabet” Biochimie vol.93,pp. 1434-1445, 2011. |
| [8] | Kleywegt G, “Use of non-crystallographic symmetry in protein structure refinement”, Acta Crystallographica D, Biological Crystallography, vol.52, no.4, pp.842–57, 1996. |
| [9] | Ye Y and Godzik A, “Flexible structure alignment by chaining aligned fragment pairs allowing twists”, Bioinformatics, vol.19, no.2, pp.ii246-ii255, 2003. |
| [10] | Jewett AI, Huang CC, Ferrin TE, “MINRMS: an efficient algorithm for determining protein structure similarity using root-mean-squared-distance”, Bioinformatics, vol.19, no.5, pp.625-634, 2003. |
| [11] | Holm L, Sander C, “Protein structure comparison by alignment of distance matrices”, Journal of Molecular Biology, vol.233, no.1, pp.123-138, 1993. |
| [12] | Dror O, Benyamini H, Nussinov R, Wolfson H, “MASS: multiple structural alignment by secondary structures”, Bioinformatics vol.19, no.1, pp.i95–i104, 2003. |
| [13] | Menke M, Berger B, Cowen L, “Matt: local flexibility aids protein multiple structure alignment”, PLoS Computational Biology, vol.4, no.1, pp.88-99, 2008. |
| [14] | Shatsky M, Nussinov R, Wolfson HJ, “A method for simultaneous alignment of multiple protein structures”, Proteins, vol.56, no.1, pp.143–156, 2004. |
| [15] | Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM, “MUSTANG: a multiple structural alignment algorithm”, Proteins, vol.64, no.3, pp.559–574, 2006. |
| [16] | Ye Y, Godzik A, “Multiple flexible structure alignment using partial order graphs”, Bioinformatics, vol.21, no.10, pp.2362– 2369, 2005. |
| [17] | Madhusudhan M,S. et al. Alignment of multiple protein structures based on sequence and structure features. Protein Engineering Design &. Selection, vol. 22, pp. 569–574, 2009. |
| [18] | Sheng W, Jian P,Jinbo X, “Alignment of distantly related protein structures: algorithm,bound and implications to homology modeling”, vol. 27, no.18,pp. 2537–2545, 2011. |
| [19] | Sali A, Blundell TL, “Definition of general topological equivalence in protein structures: A procedure involving comparison of properties and relationships through simulated annealing and dynamic programming”, Journal of Molecular Biology, vol.212, no.2, pp.403-428, 1990. |
| [20] | S. Kalaimathy, R. Sowdhamini and K. Kanagarajadurai, “Critical Assessment of Structure-based Sequence Alignment Methods at Distant Relationships”, Briefings in Bioinformatics, vol.12, no.2, pp.163-175, 2010. |
| [21] | Gandhimathi A, Nair A and Sowdhamini R, “PASS2.4: An update of database of structure-based sequence alignments of structural domain superfamilies”, Nucleic Acids Research, vol.40, no.D1, pp.D531-D534, 2012. |
| [22] | Mizuguchi K, Deane C. M, Blundell T.L, Johnson M. S and Overington J. P, “JOY: protein sequence-structure representation and analysis”, Bioinformatics, vol.14, no.7, pp.617–623, 1998. |
| [23] | Sutcliffe M. J, Haneef I, Carney D and Blundell T. L, “Knowledge based modeling of homologous proteins, Part I: Three-dimensional frameworks derived from the simultaneous superposition of multiple structures”, Protein Engineering, vol.1, no.5, pp.377–384, 1987. |
| [24] | Dinescu A, Cundari TR, Bhansali VS, Luo JL and Anderson ME, “Function of conserved residues of human glutathione synthetase: implications for the ATP-grasp enzymes”, The Journal of Biological Chemistry, vol.279, no.21, pp.22412-22421, 2004. |
| [25] | Bennett M. J, Schlunegger MP and Eisenberg D, “3D domain swapping: a mechanism for oligomer assembly”, Protein Science, vol.4, no.12, pp.2455–2468, 1995. |
| [26] | Chia-Han C,et.al, “Detection and Alignment of 3D Domain Swapping Proteins Using Angle-Distance Image-Based Secondary Structural Matching Techniques”, PLoS ONE, vol.5, no.10, e13361,2010. |
| [27] | Samuel T, Chantal Nivol,Catherine B, Marianne I,Isabelle M, Vincent M and Jean-Pierre S, “A Novel Protein Fold and Extreme Domain Swapping in the Dimeric TorD Chaperone from Shewanella massilia”, Structure, vol.11, no.2, pp.165–174, 2003. |
| [28] | Olivier G, Vincent M, Chantal I, “Multiple roles of TorD-like chaperones in the biogenesis of molybdoenzymes”, FEMS Microbiology Letters, vol.297, no.1, pp.1–9, 2009. |