-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Advances in Analytical Chemistry
p-ISSN: 2163-2839 e-ISSN: 2163-2847
2015; 5(3): 56-60
doi:10.5923/j.aac.20150503.02
A Simple Method for Determination of Chloropyramine in Tablets
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLVesna Kostik, Biljana Gjorgeska, Sofija Petkovska
Faculty of Medicine, Department of Pharmacy, University Goce Dečhev, Štip, Macedonia
Correspondence to: Vesna Kostik, Faculty of Medicine, Department of Pharmacy, University Goce Dečhev, Štip, Macedonia.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
A simple gas chromatography method with mass detection (GC-MS) for the determination of chloropyramine active pharmaceutical ingredient (API) in tablets was developed. Chromatographic separation was achieved on a fused silica ZB-5 capillary column (30 m x 0.25 mm i.d. x 0.25 µm film thickness). Acquisition was performed in selected ion monitoring mode (SIM) with target ion (58 m/z) and reference ions (71 m/z, 125 m/z).The method was validated in respect system suitability, specificity, linearity, range accuracy and precision, limit of detection (LOD), limit of quantification (LOQ), specificity, robustness and stability. The advantages of this method include simple sample treatment, short elution time (less than 15 min) and short analysis time (less than 25 min). The proposed method could be applicable for routine analysis in pharmaceutical analytical laboratories.
Keywords: Active pharmaceutical ingredient, Chloropyramine, Gas chromatography-mass detection, Tablets
Cite this paper: Vesna Kostik, Biljana Gjorgeska, Sofija Petkovska, A Simple Method for Determination of Chloropyramine in Tablets, Advances in Analytical Chemistry, Vol. 5 No. 3, 2015, pp. 56-60. doi: 10.5923/j.aac.20150503.02.
Article Outline
1. Introduction
- Chloropyramine is a first generation antihistamine drug approved in some Eastern European countries for the treatment of allergic conjunctivitis, allergic rhinitis, bronchial asthma, and other allergic conditions [1]. Chloropyramine is known as a competitive reversible H1-receptor antagonist. By blocking the effects of histamine, the drug inhibits the vasodilatation, increased vascular permeability, and tissue edema associated with histamine release in the tissue [2].Chloropyramine is an antihistamine of ethylenediamine group indicated for the treatment of several pathologies due to its anti-allergic and anti-inflammatory effects [3, 4]. This antihistamine manufactured under different trade names as Synopen®, Suprastin®, Avapena ®) etc. is still available in various pharmaceutical forms as: ointments and creams for topical application containing 0.1% (W/W) chloropyramine hydrochloride, injections containing 20 mg/mL chloropyramine hydrochloride, and tablets for oral administration containing 25 mg chloropyramine hydrochloride [5, 6]. Chloropyramine hydrochloride (CHCl) is a white powder with the chemical name(N-[(4-chlorophenyl)methyl]-N',N'-dimethyl-N-pyridin-2-ylethane-1,2-diamine hydrochloride) with the chemical formula C16H21Cl2N3 and molecular weight (average mass) 326.264 Da. The chemical structure of CHCl is shown in Figure 1.
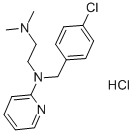 | Figure 1. Chemical structure of chloropyramine hydrochloride |
2. Materials and Methods
2.1. Reagents and Standards
- Commercially available samples, Suprastin® for oral use containing 25 mg chloropyramine in the form of chloropyramine hydrochloride were used in this study. In addition to the active ingredient, each tablet contains stearic acid, gelatine, sodium carboxymethyl starch (type A) talc, potato starch, and lactose monohydrate. Chloropyramine hydrochloride purity ≥ 99.8% was obtained from Fluka, Germany Chloropyramine base purity ≥ 99.7% was obtained from Alkaloid, Macedonia. Chloroform (Merck, Germany) gas chromatography grade was used for the extraction and preparation of standard solutions. Ammonium hydroxide (25%, V/V) was obtained from Fluka, Germany. Hydrochloric acid (37%, V/V) was obtained from Sigma Aldrich, Germany. Anhydrous sodium sulfate was obtained from Merck, Germany. Water was obtained by double distillation.
2.2. Preparation of Solutions
- Chloropyramine standard stock solution with the concentration of 1mg/mL (i) was prepared in chloroform by transferring 10 mg of chloropyramine standard accurately weighted to a 10 mL volumetric flask and filled up with chloroform to the mark. An aliquot of 1 mL of the standard stock solution (i) was transferred to a 25 mL volumetric flask and was diluted with chloroform to obtain the concentration of 40 μg/mL (standard stock solution ii).Working standard solutions, in a concentration range of 0.4 – 4.0 μg/mL, were prepared by dilution of the standard stock solution (ii) with chloroform. 1 μL of each concentration was injected into the GC-MS system. Twenty tablets were weighed and net content of each tablet was calculated. An aliquot of tablet powder equivalent to 10 mg CHCl was accurately weighed and and dissolved in approximately 10 mL 0.5 M HCl. The solution was quantitatively transferred into the 100 mL separatory funnel. pH value of the solution was adjusted to 9-10 with 25% NH4OH (V/V). After alkalization chloropyramine base was extracted with 40 mL chloroform. Chloroform layer was transferred through anhydrous sodium sulfate into the 50 mL volumetric flask and was filled up with chloroform to the mark. 0.1 mL of this solution was transferred into the 10 mL volumetric flask and filled up with chloroform to the mark. The solution was filtered through a 0.22 μm Millipore membrane filter. 2 mL of the filtrate were transferred into the auto - sampler vial. 1 μL were injected into the injection port of GC-MS.
2.3. Instrumentation and Chromatographic Conditions
- The method was performed on a Shimadzu GC-MS model QP 2010 Ultra. Chromatographic separation was achieved on a fused silica ZB-5 capillary column (30 m x 0.25 mm i.d. x 0.25 µm film thickness), supplied by Phenomenex (Torrance, USA). Helium purity 99.99999% was used as carrier gas. Operating conditions were as follows: injection port temperature, 250°C; injection volume 1 μL in the splitless mode with a sampling time of 1 min (injection pressure 250 kPa); oven temperature programme: initial column temperature was 180°C, increased with the rate of 5°C/min to 240°C and held for 5 min. The total run time of the chromatographic analysis was 14 min. The column equilibration time was 3 min, purge flow was 6 mL/min, and carrier gas constant flow was 0.62 mL/min. Ion source temperature was set at 200°C, the interface temperature was 250°C, the auxiliary temperature was 250°C, and the quadrupole temperature was 150°C.
2.4. Method Validation
- The proposed method was validated according to the guidelines set by the International Conference on Harmonization for validation of analytical procedures [12, 13]. The parameters used to validate the method of analysis were: system suitability, specificity, linearity and range, precision, limit of detection (LOD) and limit of quantification (LOQ), accuracy, robustness, and stability.
2.4.1. System Suitability
- System suitability testing is an integral part of gas chromatographic method validation performed to check and ensure on-going performance of a chromatographic system. The system repeatability was estimated by 6 repeated injections of working standard solutions at 100% of test concentration (10 μg/mL chloropyramine). The system parameters were calculated according to the recommendation of the United States Pharmacopoeia [14, 15]. The limits for system suitability parameters suggested in the FDA guideline on ״Validation of Chromatographic Methods״ [16] were used as acceptance criteria.
2.4.2. Specificity
- Specificity is the ability to asses unequivocally the analyte in the presence of components which maybe expected to be present (impurities, products of degradation, matrices, etc.) [14, 15]. Specificity of the proposed was demonstrated by spiking the grounded tablet formulation with a known quantity of CHCl standard, then checking for the interference with the placebo.
2.4.3. Linearity and Range
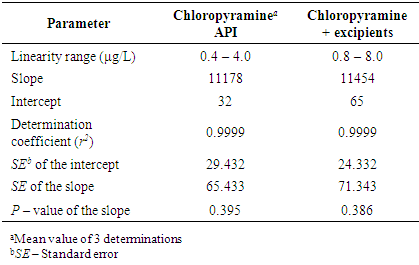
- For evaluation of linearity the calibration curve was obtained at 10 concentrations levels of chloropyramine standard solutions (0.4 – 4.0 μg/mL). Additionally, to improve the specificity, we evaluated linearity of chloropyramine in the presence of excipients. For that purpose, we spiked placebo samples with one stock active solution at six concentration levels (0.8– 8.0 μg/mL). The solutions (1 μL) were injected in triplicate into a chromatographic system with the previously given chromatographic conditions. For evaluation of linearity, the peak area and concentrations were subjected to a least square regression analysis in order to calculate calibration equation and determination coefficient.
2.4.4. Precision
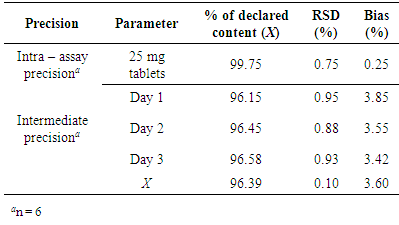
- The precision of the analytical procedure (intra-assay precision) was investigated by analyzing six sample solutions obtained by multiple sampling of the same homogenous sample under the prescribed conditions (at 100% of the test concentration of chloropyramine (10 μg/mL) on the same day, by the same analyst and using the same equipment. The intermediate precision of the analytical procedure was investigated by analyzing sample solutions on three consecutive days. The precision of the analytical procedure was expressed as the relative standard deviation of a series of measurements.
2.4.5. Limit of Detection and Limit of Quantification
- Limit of detection (LOD) and limit of quantification (LOQ) of the proposed method were determined by consecutively injecting low concentrations of the standard solutions (0.02 – 0.2 μg/mL) using the proposed GC–MS method. LOD and LOQ were calculated in accordance with ICH guidelines [14, 15] as follows: LOD = 3.3 SD/S and LOQ = 10 SD/S, where SD is the standard deviation of the response (y intercept) and S is the slope of the calibration curve obtained.
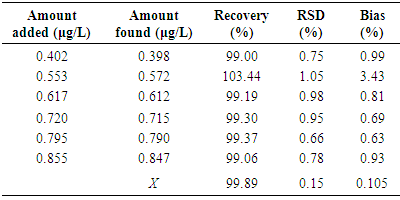
2.4.6. Accuracy
- To study the accuracy of the proposed analytical method, recovery tests were conducted using the standard addition method. To discover whether excipients interfered with the analyte, known amounts of standard were added to grinded tablet formulation samples and the resulting mixtures were analyzed by the proposed methods. The percent of recovery was calculated as follows:
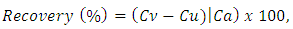 | (1) |
2.4.7. Method Robustness
- The capacity of the proposed method to remain unaffected by small (deliberate) variations in parameters was evaluated in order to determine method robustness. Changes were made to the following method parameters: concentration of HCl (± 10%), pH value of the solution after addition of 25% NH4OH (± 5%), and temperature (± 3%).
2.4.8. Stability of the Sample Extracts
- For this study five quality control (QC) samples with the nominal concentration of 1 mg/g CHCl were used. After the sample preparation according to the procedure described above (2.2), obtained extracts were stored at -20°C over the period of 12 weeks. The concentration of chloropyramine was determined according to the procedure described in 2.3.
3. Results and Discussion
3.1. Method Optimization
- Because of poor solubility of chloropyramine hydrochloride (salt) in water and in the majority of organic solvents (polar and non-polar), in our experiment as a suitable solvent for chloropyramine hydrochloride dissolution we used 0.5 M HCl. Iliaszenko et al. used 0.5 M HCl for dissolution of several tertiary amine hydrochloride drugs in coated tablets, prior their spectrophotometric determination [7]. Chloropyramine base was deliberated from the salt by addition of 25% NH4OH (V/V) to pH 9-10. Extraction of the free base was efficiently performed with chloroform.In order to improve specificity and selectivity and minimize interferences from ointment or solvent systems that may occur in spetrophotometric or HPLC determinations, we performed the analysis using GC-MS system. In our previous study, we use the same GC-MS technique, after the isolation of chloropyramine from ointments [17]. Selectivity was obtained by operating in selected ion monitoring (SIM) mode with target ion (58 m/z) and reference ions (71 m/z, 125 m/z). Figure 2 depicts the mass spectrum of chloropyramine.
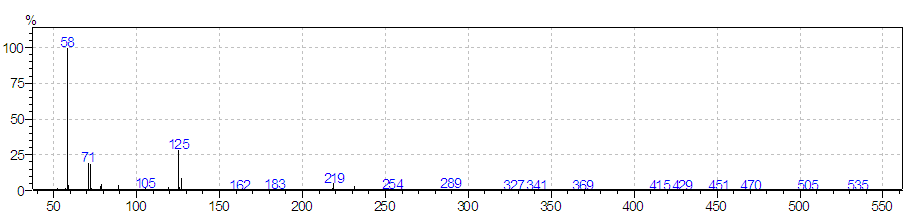 | Figure 2. Mass spectrum of chloropyramine |
3.2. Method Optimization
3.2.1. System Suitability
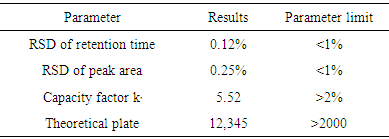
- The results of system suitability test were found within the acceptable range [16] indicating that the system was suitable for the intended analysis (Table 1).
|
3.2.2. Specificity
- In the specificity study, standard solutions of chloropyramine and the sample solution were injected and a single peak was obtained for chloropyramine, which indicates that that there was no interference from the excipient used or from the chemicals.
3.2.3. Linearity and Range
- In the present study, linearity was studied in the concentration range 0.4 – 4.0 μg/mL chloropyrmine and the following regression equation was found by plotting the peak area (y) expressed in AU versus the chloropyramine concentration (x) expressed in μg/L: y = 11178 x + 32 (r2 = 0.9999). The determination coefficient (r2) demonstrates the excellent relationship between the peak area and concentration of chloropyramine. The calibration curve of chloropyramine API and chloropyramine with placebo were linear. The regression lines were not significantly different from a curve passing through the origin y = a x. (P-value of the slope > 0.05). The excipients had no influence and there was no matrix effect observed (Table 2).
|
3.2.4. Precision and Accuracy
- The variation in results obtained within a day (RSD = 0.75%, for Suprastin® 25 mg tablets) and day to day variations (RSD = 0.10%) for chloropyramine determination was very low (≤ 2%), which confirmed the precision of the method. The statistical data are shown in Table 3.
|
|
3.2.5. Limit of Detection and Limit of Quantification
- The limit of detection (LOD) and limit of quantification (LOQ) were 0.0132 μg/mL (corresponding to 0.0132 ng/mL loaded onto column) and 0.04 μg/mL (corresponding to 0.04 ng/mL loaded onto column), respectively.
3.2.6. Robustness
- Based on the obtained results from the method robustness (RSD < 2%), the proposed GC-MS analytical method were demonstrated to be robust (Table 5).
|

3.2.7. Stability
- The results of our investigations showed that the extracts were stable at -20°C within a period of 4 weeks. Approximate degradation observed for 4, 8 and 12 weeks was 1.25%, 67.8% and 77.4%, respectively. Figure 3 shows chromatograms of extracts of chloropyramine base in chloroform over the period from 4 to 12 weeks.
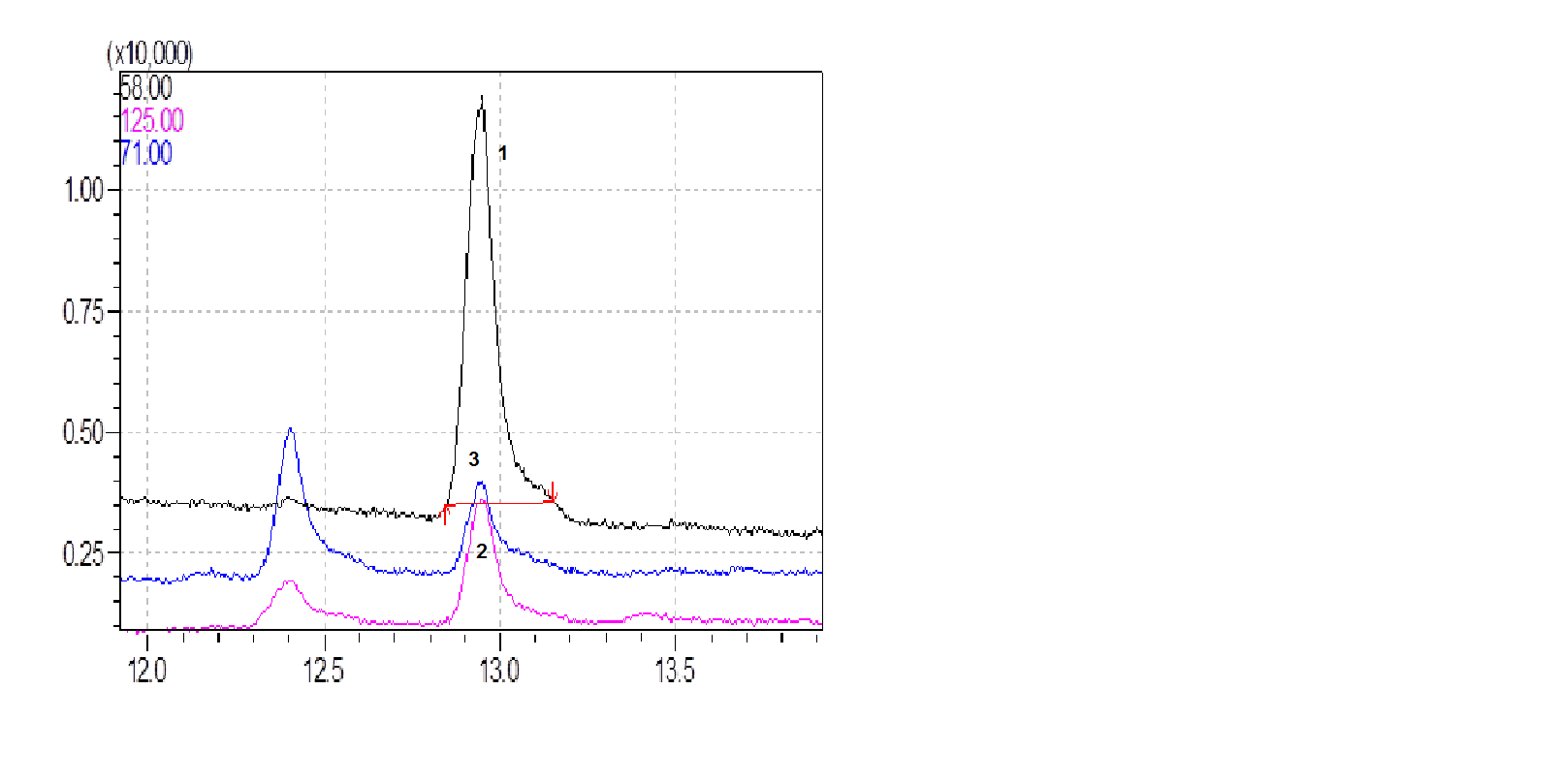 | Figure 3. Overlay of chromatograms of extracts of chloropyramine base in chloroform (1-after 4 weeks, 2-after 8 weeks and 3-after 12 weeks of storage at -20°C) |
4. Conclusions
- The proposed GC-MS method allows a simple, accurate, precise and rapid determination of chloropyramine API in tablets. The important features and advantages of this method include: simple sample extraction of small amounts of powdered sample at ambient temperature, relatively short elution time (less then 13 min) and shorter analysis time than in previously reported methods (less than 25min); good precision (RSD less than 2%) and high recovery (higher than 99%). Furthermore, operating in SIM mode provides additional selectivity of the method in terms of interferences. The proposed method could be applicable for routine analysis in pharmaceutical analytical laboratories.