-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Advances in Analytical Chemistry
p-ISSN: 2163-2839 e-ISSN: 2163-2847
2013; 3(4): 48-53
doi:10.5923/j.aac.20130304.02
End of Supply Chain Screening to Assess Drug Product Quality in the U.S. Market – Omeprazole as a Case Study
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLRichard T. Addo1, E. Blake Watkins1, Joel S. Owen1, Kenneth R. Davis1, Ronald C. Bingham2
1Department of Pharmaceutical Sciences, School of Pharmacy, Union University, Jackson, TN, 38305
2Physical Medicine and Rehabilitation, Jackson, TN, 38305
Correspondence to: Richard T. Addo, Department of Pharmaceutical Sciences, School of Pharmacy, Union University, Jackson, TN, 38305.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Drug products in the U.S. market are tested prior to shipment from sites of manufacture, but limited or no testing is required once products are shipped from the site of manufacture before reaching the patient. In this era of worldwide manufacture and distribution considerations of product quality are raised. Recent episodes of counterfeiting and the possibility of substandard and degraded product bring such considerations to the forefront. This work selected a target active pharmaceutical ingredient, omeprazole, and tested each of the orally administered products available on the U.S. market at a snapshot in time for compliance with the USP release requirement for drug content. Twenty distinct omeprazole products representing each of the approved formulations for marketing in the U.S. were purchased through a major wholesaler or local pharmacy and tested for drug content. Drug was extracted from the dosage form using ACN:Methanol (1:1) following removal of the enteric coating from beaded products using NaOH (0.1 N). The assay was performed using a UPLC with scanning UV detection from 210-400 nm. Omeprazole content in every product fell within the USP requirements of 90% to 110% of label claim. The maximum product strength observed was 102.09% of label claim, and the minimum was 92.92%. This snapshot of the quality of omeprazole product on the U.S. market reflects robust product quality for both innovator and generic products, and for products manufactured in the U.S. and overseas.
Keywords: Drug Product Quality, Omeprazole, UPLC
Cite this paper: Richard T. Addo, E. Blake Watkins, Joel S. Owen, Kenneth R. Davis, Ronald C. Bingham, End of Supply Chain Screening to Assess Drug Product Quality in the U.S. Market – Omeprazole as a Case Study, Advances in Analytical Chemistry, Vol. 3 No. 4, 2013, pp. 48-53. doi: 10.5923/j.aac.20130304.02.
Article Outline
1. Introduction
- Patients with serious or chronic diseases such as epilepsy, cancer, infections, hypertension, or Parkinson’s Disease deserve to receive effective medicines. To have confidence in the medicines they consume, patients and pharmaceutical product consumers in the U.S. depend upon the good reputation of the practice of pharmacy and of the historical quality of pharmaceutical products. However, many news reports in recent months and years regarding product recalls, tampering, fraud, and counterfeiting give appropriate cause for patient concern regarding the quality of products they consume. Both drug manufacturers and FDA work diligently to ensure the quality of products manufactured, but there is significant space for wrongdoing and poor product control within the complex supply chain that provides products to patients after they have shipped from the site of manufacture. There is no consistently applied testing of pharmaceutical products once they have shipped from the site of manufacture.Current federal regulations governing the manufacture and marketing of drug products in the United States require that each lot of a pharmaceutical product be tested prior to shipment from its site of manufacture and that retention samples from each lot be tested periodically throughout the full shelf-life of the lot.[1] Unfortunately, most products are not tested at any point following shipment from the site of manufacture until they reach the patient. When a product arrives at a pharmacy from a legitimate, licensed distributor and the packaging appears to be in original condition, the pharmacist dispensing the medication must assume as a routine element of practice that the product meets its regulatory standards of quality and that those standards are sufficient for adequate safety and efficacy in the treatment of the patient.The U.S. pharmaceutical product market is considered a closed system, with control of sources and shipments of products between licensed entities. However, reports of fraud and the growing worldwide manufacturing of products marketed in the U.S. accentuate the possibility that product quality may occasionally be lacking. A recent FDA announcement of the distribution of counterfeit Avastin® to more than 20 states brings this to the forefront as a current, local, and vital issue of public safety.[2] The scope of fraudulent activity in this case was broad, involving an elaborate network of companies suggestive of organized crime.[3]Poor quality medications can be classified into three key categories: counterfeit, substandard, and degraded.[4] Counterfeit medications are produced with intentional fraudulent activity and are “misbranded” with intent.[5] Counterfeiting of medications is clearly a criminal act. Congress has recently proposed in H.R. 3468 the strengthening of penalties for individuals convicted of counterfeiting with fines up to $4,000,000 and life imprisonment.[6] One individual was recently sentenced by the U.S. federal court in Miami to 4 years in prison, $25,000 in fines, and the forfeiture of $300,000, for conspiracy to commit mail fraud in connection with the sale of foreign and counterfeit medicines to U.S. customers.[7]Substandard products may occur for many reasons including inadequacies in raw material testing, poor conditions and procedures of manufacture and control, inadequate training of production workers, or poor maintenance of production equipment. Inadequate quality procedures may lead to the unintentional release of substandard product onto the market. Intentional fraudulent activity may also result in the release of substandard product. In either case, a substandard product is defined by FDA as misbranded. FDA oversight is a key component of enforcing the adherence to quality standards for products marketed in the U.S.A. Where oversight is lacking, there is a greater opportunity for substandard products to emerge, which is true of both innovator and generic companies.Some drug products are initially produced with adequate quality, but later in time lack conformity to product quality standards due to the effects of time and the environment. Such products are considered degraded. If tested, a degraded product would no longer meet the release specifications of the original product. Poor shipping and storage conditions may contribute to the degradation potential of drug products. The effects of temperature and humidity are of particular concern. Products arriving in the U.S. market from distant regions of the world have a significant opportunity for degradation to occur if shipment conditions are not properly controlled.Pharmaceutical manufacturing in countries outside of North America has increased dramatically in recent years, including countries with totalitarian governments and limited infrastructure for the control and maintenance of records and products.[8] According to U.S. Representative John D. Dingell (MI), a senior member of the House Committee on Energy and Commerce that has jurisdiction of the FDA, nearly 40% of our drug products and 80% of drug ingredients are produced overseas.[9] Janet Woodcock, MD, Director of the Center for Drug Evaluation and Research of the FDA reported to Congress that there are currently more drug manufacturing facilities overseas to be inspected than domestic facilities.[10] In a recent Government Accounting Office publication the FDA acknowledged the difficulty of performing inspections abroad. The ability to perform unannounced inspections is logistically difficult, and the FDA lacks the authority to force companies in sovereign nations to allow unimpeded inspection of manufacturing facilities and records.[10] It is estimated that in 2009 the FDA inspected 9% of foreign establishments subject to inspection, in contrast to 40% of domestic establishments in the same year. The dramatic increase in manufacturing outside the US and the difficulty for proper oversight of production and the supply chain provide cause to consider the safety of our drug supply. Significant moves have been made or are planned to strengthen FDA inspection rates and increase access overseas, including increased funding for foreign inspections, collaboration with inspection activities of other regulatory authorities around the world, the establishment of an inspection office in China, and the use of a risk-based inspection approach.[11]The pharmacy supply chain in the U.S. is a complicated system with many participants, including manufacturers, distributors, repackagers, hospitals, retail outlets, pharmacists, doctors, and nurses. A 2001 report to Congress provided a statistical profile of wholesalers.[12] The report described 6,500 licensed wholesalers, of whom 5 major wholesalers control 90% of the market in the U.S. Many of the smaller wholesalers specialize in certain product types or regions in the country.The FDA Office of Drug Integrity and Supply Chain Security works to monitor and ensure the quality of drug products presented to U.S. consumers using risk- and science - based policy development, surveillance, and enforcement. Though government and private organizations[13] exist that seek to maximize control of the U.S. pharmaceutical product supply chain in a cost effective manner, these are not completely adequate to ensure the safety of the public from intentional or unintentional exposure to poor quality medicines. Perhaps no organization could achieve that goal, especially with the advent of internet pharmacy and international product manufacturing and distribution.With recent reports of fraudulent and counterfeiting activity in pharmaceutical products, and with the potentially increased likelihood of drug degradation during prolonged shipment from an increasing number of manufacturing sites around the world, testing marketed products seems a reasonable measure to evaluate the frequency of problems and risk to the public.
 | Figure 1. Chemical structure of omeprazole |
|
2. Methods
2.1. Product Acquisition
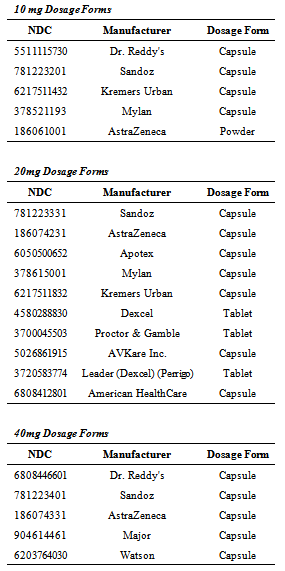
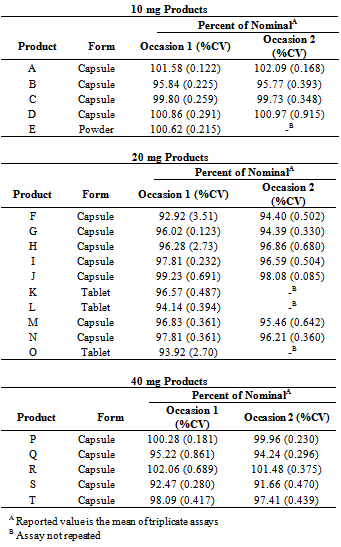
- Approved NDA and ANDA products were identified on the FDA database of approved NDAs at Drugs@FDA.[15] Each of the currently marketed, orally administered omeprazole products and strengths was purchased through one of the major wholesalers in the US market or from a local, retail pharmacy setting. Both innovator and generic products were purchased, as well as OTC and prescription products. Dosage forms included tablets, capsules, and a powder for reconstitution. Four 10-mg capsule products and one 10-mg powder were tested. Seven 20-mg capsule products and three 20-mg tablet products were tested. Five 40-mg capsule products were tested. In total, 20 marketed products of omeprazole were tested. The history of purchase location or supplier with receipts or invoices was maintained in the study records. Product name, strength, dosage form, manufacturer, lot number, package quantity, and quantity purchased were recorded.The products tested are listed in Table 1. A sufficient quantity of each product was purchased to allow completion of the extraction and assay procedures on three occasions. Unused samples for retention are maintained at the Union University School of Pharmacy in controlled room temperature or refrigerated conditions.
2.2. Analytical Methods
- Omeprazole (USP standard) was obtained from Sigma Aldrich (St. Louis, MO, USA). Omeprazole (10 mg) was weighed on a certified, calibrated analytical balance and dissolved in 50 mL of 1:1 acetonitrile:HPLC water to give a 200 µg/mL working standard. Acetonitrile (ACN) and HPLC water were obtained from VWR (Radnor, PA). The working standard solution was immediately refrigerated. Prior to UPLC analysis, samples were filtered through a 0.2-µm filter. Analysis of samples was performed on a Waters (Milford, MA, USA) Acquity H-class ultra high performance liquid chromatography (UPLC) system equipped with a quaternary solvent delivery system, a refrigerated sample manager, a column heater, a photo diode array detector scanning from 210-400 nm and with a Waters BEH C18 (50 mm x 3.0 mm, 1.7 µm particle size) analytical column with a Waters BEH C18 (2.1 mm x5 mm, 1.7 µm) guard column.The UPLC injection volume was 2 µL, with a flow rate of 0.45 mL/min. Omeprazole exhibits a λmax at 300.8 nm. The retention time was 1.57 minutes for omeprazole. A standard curve was developed with stock solutions of 10, 25, 50, 75, 100 and 150 µg/mL, which were prepared by the dilution of a 200 µg/mL stock solution with 1:1 ACN:Methanol.The assay was linear (r2 > 0.998) over the range tested (10-µg/mL to 200-µg/mL). The intra- and inter-day variation was 0.7 % RSD and 4.0 %RSD, respectively.Analytical data were captured electronically using Masslynx 4.1 (Waters, Milford, MA). Statistics were computed using Microsoft Excel 2010 (Redmond, WA, USA).
2.3. Sample Analysis
- Marketed product samples were prepared for analysis using USP guidelines, but with some modifications as described here. Twenty (20) capsules were weighed together and individually for each product. The powder content was separated from each capsule, and both the powder and capsule shells were weighed separately. For the powder product, the powder was removed from the foil package and weighed. The average powder weight was computed for each product. The powder from all the units of a product was blended in a mortar and an amount to contain the labeled amount of drug was removed and weighed precisely.Extraction of drug from the product was performed by first removing the enteric coating from the beads using 1 mL of 0.1 N NaOH for approximately 5 minutes. ACN (50%) was added to dissolve the bead or tablet content with vortexing for a minimum of 15 minutes, until completely dissolved. Samples were brought to the appropriate volume with 50% ACN and filtered through a 0.2-µm filter prior to analysis. Samples from the powder formulation did not completely dissolve. The samples were centrifuged at 9,000 rpm for 5 minutes at 21℃. The supernatant was filtered for UPLC analysis.
3. Results
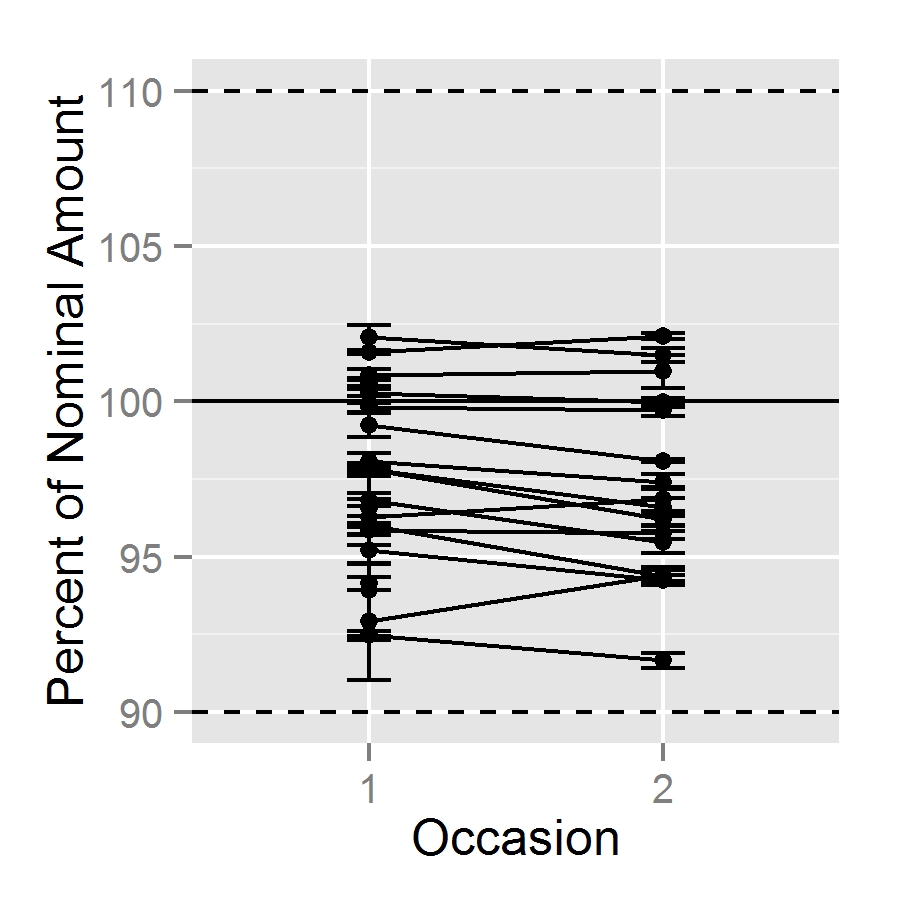 | Figure 2. Assay results by occasion of assay, connected by product, with Standard Error |
|
4. Discussion
- The present work represents a snapshot of the market, in a limited region, in a brief instance of time, for a single product. Thus, this work does not necessarily reflect the overall pharmaceutical product market. We believe that further work should be done to evaluate the quality of all marketed products at the end of the supply chain. Even so, execution of the project was expensive in both resources and time. The expense was partly related to the cost of the products selected, but this will be the case for many pharmaceutical products, and it is likely that more expensive products will be more attractive targets for counterfeiting. A system of routine testing of all products and lots as they move through the supply chain to the consumer would likely be prohibitively expensive. Screening approaches might be appropriate to take a pulse of the quality of products on the market. If routine screening of products in the marketplace were to be instituted, a reimbursement system by product manufacturers might be considered. In the interest of consumer confidence, manufacturers might be interested in reimbursing this cost of medication and testing of their products.However, obtaining the samples to be tested should be a random process after the product leaves the site of manufacture. The purchase of products from stores or wholesale distributors, close in the supply chain to patient delivery, gives the best representation possible of product actually delivered to patients without obtaining products directly from patients.Obtaining drug products for testing requires appropriate licensing, accounts, or legal procedures for access. A university or research lab may have a license with their state board of pharmacy enabling them to set up accounts with wholesalers, but the ability to acquire drug products directly from a pharmacy will vary by state law. A pharmacy typically sells prescription drug products to a patient in response to a prescription, as part of the doctor, patient, and pharmacist triad. State laws may vary, but there are processes of transferring drug products between licensed entities “by invoice” or similar procedures. The method of sample collection does depend upon the question being asked and influences the conclusions that can be drawn.[4] Careful consideration of the method of sampling and the question of interest are important steps in designing any such screening program. The current study collected samples from all products and strengths of the approved manufacturers of orally administered omeprazole in the U.S. Sampling was limited in time and location, though the majority of the products were obtained from one of the major wholesalers in the U.S.When obtaining samples for screening, sufficient sample should be acquired to allow the retesting of the product if aberrant results are found. The retention samples should be maintained appropriately and with sufficient documentation to account for their acquisition and storage. If out – of - specification results are found, there is at least a moral obligation of the testing entity to notify both the manufacturer and the FDA of this finding. Having sufficient sample quantity for retesting will aid in the resolution of disputes.As a matter of public safety, establishing a screening program to test marketed products seems a reasonable measure to evaluate the frequency of problems and risk to the public. The frequency and independence of testing must, however, be balanced against the cost and resources for performing such analyses. Appropriate statistical sampling considerations, product acquisition methods, laboratory testing methods, financing, and reporting requirements must be considered.
5. Conclusions
- In this study, each of the marketed omeprazole products assayed met the USP requirements for content of active pharmaceutical ingredient. Thus, this snapshot of the quality of product on the U.S. market reflects robust product quality for both innovator and generic products, and for products manufactured in the U.S. and overseas.
ACKNOWLEDGEMENTS
- All funding for this project was provided by Ronald C. Bingham, M.D., Jackson, TN, who is one of the authors of this manuscript. The authors wish to gratefully acknowledge the work of Amanda Casey in assisting with administrative aspects of this research project.