-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Advances in Analytical Chemistry
p-ISSN: 2163-2839 e-ISSN: 2163-2847
2013; 3(4): 41-47
doi:10.5923/j.aac.20130304.01
The Preparation of Iron(II) Complex from Iron(III) Chloride, under Aerobic Conditions
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLM. A. Z. Eltayeb, N. M. Modawe
Department of Chemistry, Faculty of Science and Technology , Al-Neelain University, Khartoum, Sudan
Correspondence to: N. M. Modawe, Department of Chemistry, Faculty of Science and Technology , Al-Neelain University, Khartoum, Sudan.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
The reaction of iron(III) chloride with Schiff base (DTOH2) formed from the condensation of diacetylmonoxime and thiosemicarbazide under aerobic conditions affords exclusively the iron(II) complex. The ligand acts both as a reducing and a complexing agent. This is supported by the yield and by the oxidation product of the ligand. The reducing capacity of the DTOH2 ligand prevents the formation of iron(III) complex. The iron(II) complex was characterized by IR, NMR and Mössbauer spectroscopy and possesses a distorted octahedral coordination geometry. The iron(II) complex is diamagnetic suggesting a low spin state. In the iron(II) complex the ligand, DTOH2, behaves as a tridentate ligand and is coordinated through the azomethine nitrogen, oxime nitrogen and the thione sulfur.
Keywords: Schiff Base (DTOH2), Diacetylmonoxime, Thiosemicarbazide, Aerobic Conditions, Iron(II) Complex
Cite this paper: M. A. Z. Eltayeb, N. M. Modawe, The Preparation of Iron(II) Complex from Iron(III) Chloride, under Aerobic Conditions, Advances in Analytical Chemistry, Vol. 3 No. 4, 2013, pp. 41-47. doi: 10.5923/j.aac.20130304.01.
Article Outline
1. Introduction
- The chemistry of thiosemicarbazones chelating ligands has attracted considerable interest primarily due to their broad biological activity, for instance antibacterial, antifungal, and anti HIV[1] as well as their metal chelating abilities and reductive capacity[2, 3]. The π delocalization of charge and the configuration flexibility of their molecular chain can give rise to a great variety of coordination modes. The synthesis and characterization of iron complexes with Schiff – base ligands play a relevant role in the coordination chemistry of iron due to their importance as synthetic models for the iron – containing enzymes[4,5,6] . In addition, the understanding of the factors which control the spin state of the iron ion with these ligands require more attention.[7,8]. The reaction conditions, nature of ligand donor set that yield a stable metal complex in its + 2, +3 or +4 oxidation state are yet not clear[9, 10]. Some studies have been carried out mainly to characterize the products of the reactions between Nx Sy – containing ligands and Fe(III) ions under aerobic conditions. The mixtures of Fe(III) and 2,4,6,-tri(2-pyridyl) -1,3,5,triazine(tptz) in the presence of benzilic acid resulted in partial and / or quantitative metal reduction[11]. Replacement of the oxidative NO3- ions of Fe(III) salt with the innocent ClO4- ions in the reaction mixture, resulted in quantitative reduction of Fe(III) yielded exclusively the low spin hexacoordinate Fe(II) complex[Fe(II)(tptz)2](ClO4)4. Raper[12], reported the first Fe(III) promoted in situ oxidation of a thiozoline-2-thione to the corresponding hetero-disulphide with concomitant coordination to Fe(II). The acidic solution of Fe(III) – indole-3-butric acid kept at ambient temperature under aerobic conditions resulted in the reduction of 85 % of Fe(III) within 48 h[13]. The aerobic reaction of Fe(III) chloride with the Schiff base formed from the condensation of tren and either 2-pyridine carboxaldehyde or 1-methyl-2-pyridine carboxaldehyde offords exclusively the Fe(II) tripodal complexes. The complexes are low spin and spin crossover respectively[14]. The addition of preformed Schiff base ligands (chelating ligands with imine nitrogen atoms and thiolato sulphur atoms) to Fe(III) salts often results in the reduction and formation of Fe(II) complexes and/or S-bridged polymeric species[15]. In general, syntheses of Fe(III) complex with Fe(III) – S (thiolato) bonds beset with problems of oligomerization and/or autoredox reactions generating Fe(II) and RSSR[16, 17]. Diacetylthiosemicarbazoneoxime can exist in two tautomeric forms A and B (scheme 1). In form A it can act as a tridentate ligand by coordinating either through the oxime N and by imino N and thiocarbonyl S or as mononegatively charged tridentate ligand by losing a proton from the mercapto group of the tautomeric form B .In this contribution we report the synthesis and physicochemical studies of iron –diacetylthiosemicarbazoneoxime complex.
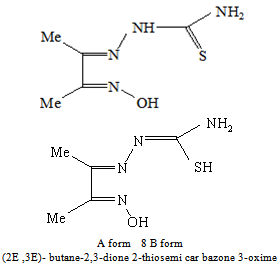 | Scheme (1). Tautomeric forms A and B of Diacetylthiosemicarbazone |
2. Experimental
- All chemicals and solvents used in the preparation of the ligand and complex were of reagent grade and were used as received. All manipulations were carried out under aerobic conditions. The ligand was synthesized using the published method[18].
2.1. Syntheses
2.1.1. Diacetylthiosemicarbazoneoxime ,[DTOH2]
- A 2, 3-butanedione monoxime (8.10 g, 80 mmol) and thiosemicarbazide (7.30 g, 80 mmol) were mixed in 85% ethanol/water (250 mL). The mixture was boiled under reflux with stirring for 4 h. The solution volume was reduced to half its original volume. The resulting white precipitate was collected by suction filtration and washed with cold ethanol. The crude product was crystallized from ethanol as white crystalline plates. Yield: 12.5 g (90 %), m. p. 200℃ (literature value 200.2℃). Anal. Caled: for C9 H10 N4 O S , M = 174 g/mole: C, 34.50; H, 5.74; N, 32.16. Found: C, 34.52; H, 5.81; N, 31.95. Selected IR bands (KBr pellet, cm-1): corresponding to ν(OH), ν(NH), ν(NH2), ν(C=N), ν(C=S), at 3411(s); 3233(s); 3144 and 3050 doublet; 1600(s); and 839(m), respectively. 1H NMR (DMSO, 500 MHz), δ from TMS: 2.06 (d, CH3), 8.02 (d, NH2), 10.18 (s, NH), 11.54 (s, OH).
2.1.2. Iron Complex[Fe(II) (DTOH2)2]Cl2 (I)
- A solution of FeCl3.6H2O (1.35 g, 5 mmol) in EtOH (20 mL) was added to a warm solution of DTOH2 (1.76 g, 10 mmol) in EtOH (20 mL). The mixture was boiled under reflux for 0.5 h. The resulting diamagnetic dark red precipitate was filtered off, washed several times with cold EtOH and Et2O and dried in vacuu, (yield; 0.95 g 40%). Anal.Caled for C18H20N8O2S2FeCl2, M = 474.8 g/mole: C, 25.27; H, 4.21; N, 23.58; S, 13.50; Fe, 11.79. Found; C, 25.21; H, 4.45; N, 22.89; S, 14.45; Fe, 11.80.Selected IR bands (KBr pellet, cm-1): corresponding to ν(OH), ν(NH), ν(NH2), ν(C=N), ν(C=S), at 3415(w); 3253(s); 3131 and 3050 doublet; 1615 and 1563 doublet; and 828(w), respectively. 1H NMR (DMSO, 500 MHz), δ from TMS: 2.02 (d, CH3), 8.09 (d, NH2), 10.00 (s, NH), 11.55 (s, OH).
2.2. Physical Measurements
- IH-NMR. spectra were recorded on a Varian spectrometer using DMSO as solvent. Chemical shift (δ) are reported in p.p.m. relative to Me4Si. C, H, N, and S contents were determined at UMIST, Manchester (UK), Fe was determined by pyrolysis of the solid complex at 600℃ and then weighing the metallic oxide residue[19]. The IR spectra were obtained on Nicolet 51 OP-FT-IR spectrometer with samples prepared as KBr pellets. Magnetic susceptibility measurements were carried out at room temperature using an Evan balance. Molar conductivities (0.001M in H2O) were measured with CMD 8500 Laboratory Conductivity Meter. Thermogravimetric data were obtained on Perkin–Elmer TGA7 unit (heating rate 10℃/ min). Cyclic voltammetry was performed in DMSO (Aldrich) with a supporting electrolyte of (nBu4)(PF6) (0.1 M Fluka) with a potentiostat / wave generator (Oxford Electrodes) equipped with an Allen X-Y recorder. The electrochemical cell contained a Pt wire working electrode, an Ag wire a quasi-reference electrode, and a Pt wire auxiliary electrode. The voltammetric data are compared with (Fc/ Fc)0/+, E = + 0.44 V and ΔE = 70 mV for the cell used. Formal potentials were calculated as the average of anodic and cathodic peaks potentials and are reported versus NHE. Mössbauer investigations were performed at MPI für Bioanorganische Chemie/ Mülheim by using a constant acceleration spectrometer. Resonance Raman spectroscopy was obtained on IRA300 Laser Raman Spectrometer- Lambda Scientific Pty with sample prepared by mixing the sample in the KBr matrix in tightly covered NMR tubes.
3. Results and Discussion
- The Schiff-base ligand DTOH2 was synthesized with some modification of the original procedure[18] and coordinate to Fe(II) ion as tridentate ligand in its neutral A form. As shown in scheme 1, the ligand employs one oxime nitrogen, one imine nitrogen and one thione sulfur to bind iron(II) in this complex. The iron(II) complex was synthesized in low yield by the reaction of FeCl3.6H2O with DTOH2 in a molar ratio 1:2 using ethanol as solvent and with reflux. The low yield of the compound is attributed to the participation of the DTOH2 ligand in redox reaction with Fe(III) ion, where Fe(III) ion was reduced to Fe(II) ion. The Fe(II) ion produced was complexes by the remaining DTOH2 ligand. In order to verify the quantitative reduction of Fe(III) ion by this ligand we have carried out certain "blind" experiments. Our first attempt involved the reaction of 1 equivalent FeCl3.6H2O with 3 equivalents DTOH2 in order to increase the yield assuming that one DTOH2 molecule offers one electron, which afforded 85% of the Fe(II) complex. Our next attempt involved the examination of the possible product from the oxidation of the ligand in order to verify the role of the reductive and chelating ligand (these procedures are not described in the Experimental part). The filtrate from the reaction of Fe(III) ion and DTOH2 was extracted by diethyl ether and the solid left after the evaporation of the ether was compared with the authentic sample prepared by the oxidation of the ligand by iodine. On the basis of Raman spectra (Fig.1) which indicated the presence of disulfide bond in both samples at 482.0 cm-1 (oxidation product of the filtrate ) and 471.9 cm-1 (oxidation of the authentic sample by iodine) peak 2 on the two spectra respectively, we concluded that the ligand acts both as a reducing and as a complexing agent. Therefore, with this ligand it is not possible to prepare the iron (III) complex by direct reaction. The Fe(II) complex is stable in the solid state as well as in solution at ambient temperature. The complex is soluble in H2O, EtOH and DMSO. The complex has been formulated as [Fe(II)(DTOH2)2]Cl2 on the basis of its elemental , spectroscopic (IR,, IH-NMR, Mőssbauer) data, conductivity and magnetic susceptibility measurements and thermogravimetric study, and is shown in scheme 2. This structure is similar to that previously prepared using FeCl2[20]. The elemental analyses are consistent with the formula,[Fe (DTOH2)2.Cl2]. The conductivity of the iron(II) complex was determined as 250 Ω-1 cm2 mol-1. This is consistent with formation of a 1:2electrolyte and indicates that the ligand coordinated to the Fe (II) in the neutral form. This is in agreement with the I H NMR, IR spectra and Mössbauer data of the complex.
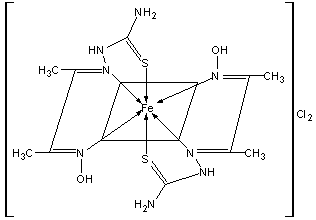 | Scheme 2. Structure of the Iron(II) complex |
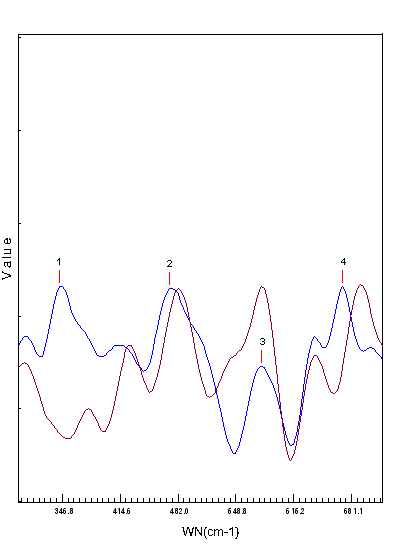 | Figure 1. Raman spectra of oxidation product of the ligand by Fe(III) (red color) and the authentic sample by iodine (blue color) |
3.1. Spectroscopic Studies
- The IR spectrum of the ligand (Figure 2 a) in its uncoordinated form shows bands in the 3400-3100 cm-1 region which are attributable to stretching modes of OH, NH, and NH2 groups. These bands are also present in the IR spectrum of the complex (Figure 2 b) with very small shifts. This confirms the coordination of the ligand to the metal in its neutral form. The absence of the υ (SH) band and the presence of the υ (NH) as well as the characteristic "thioamide" bands confirms the presence of the thione form in the ligand and the complex[21]. The diacetylthiosemicarbazoneoxime υ (C=N) unresolved band at 1600 cm-1 shifts in the complex into doublet of higher and lower wavenumber at 1615 and 1563 cm-1 indicating coordination of the oxime nitrogen and the azomethine nitrogen. The spectrum of the uncomplexed diacetylthiosemicarbazone shows the thioamide IV which possess a band at 839 cm-1. This band shifts by 11 cm-1 to lower frequencies in the complex, indicating that a thione sulfur is attached to the metal center, in agreement with previous studies of thiosemicarbazone complexes[22]. Bands in the 350 – 375 cm-1 region are assigned to υ (FeS) bonds and that in 435-480 cm-1 region to υ (FeN) vibrations[23]. This supports the coordination of the ligand via NNS.
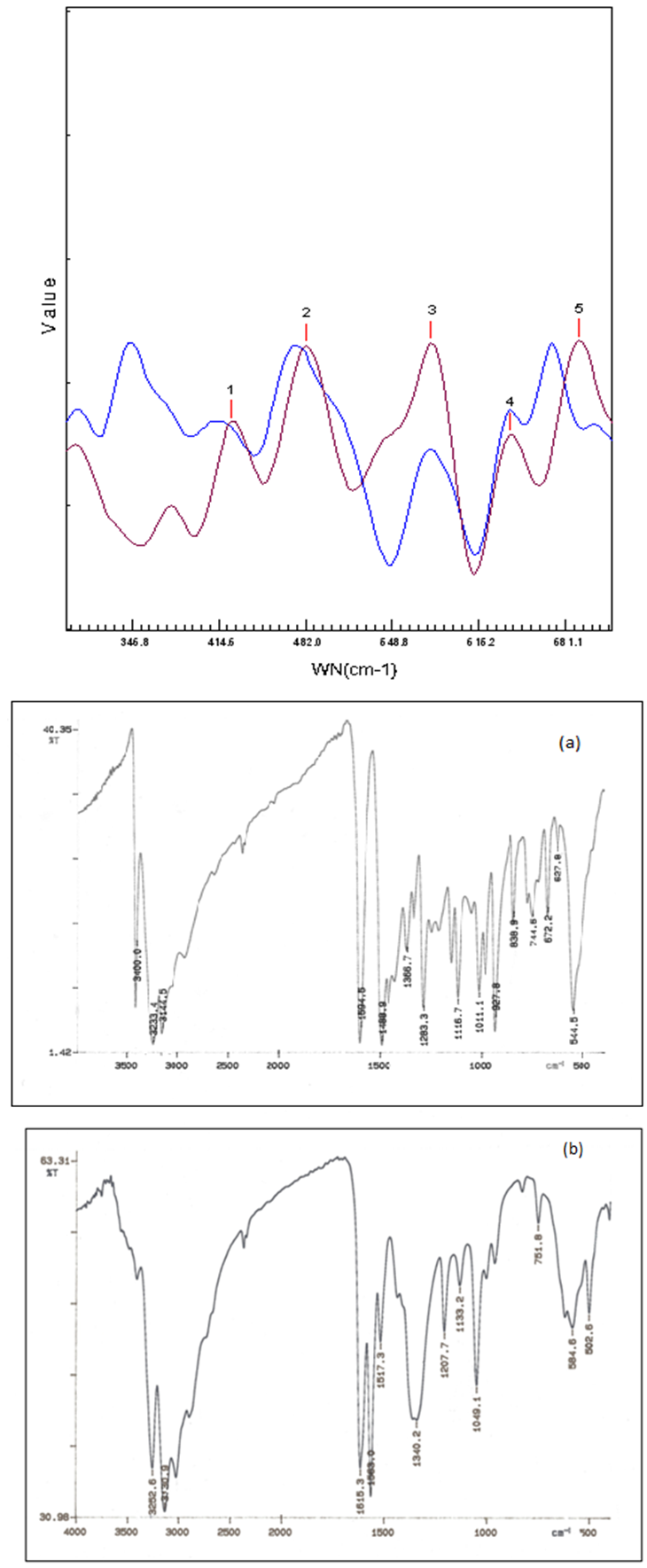 | Figure 2. Infra-red spectra from powder samples of the ligand (a) and its iron(II) complex(b) |
3.2. IH NMR Spectroscopy
- The bonding pattern of the ligand and its complex is further supported by the proton magnetic resonance spectral studies in DMSO - d6. The free ligand exhibit a singlet at δ 11.54 p.p.m. due to OH. The other singlet that appeare at δ 10.18 p.p.m.is due to NH. The ligand shows multiplets at δ 8.02 p.p.m. are attributed to the NH2 group.The doublet at δ 2.01 and δ 2.11p.p.m. is assigned to the two methyl groups. The number of protons related to the chemical shifts observed in the spectrum of the ligand remained unchanged in the diamagnetic metal complex[24]. The chemical shift δ (p.p.m) owing to acetamide (NH) and hydrazine (NH) protons remain practically unchanged in the complex, indicating neither acetamide (NH) nitrogen nor hydrazine (NH) nitrogen are involved in ligand coordination to the metal. These results confirm that the ligand is coordinated to the metal ion in its neutral form.
3.3. Mössbauer Data
- The Mössbauer spectrum at 80.0 K of iron (II) complex (I) is shown in Figure 3. Table 1 gives the experimental data as well as those of Fe(II) complex (II) of the related ligand 2-acetylpyridine thiosemicarbazone studied by other authors[25]. Complex (II) is represented by[Fe(HAPT)2]Cl2 in which HAPT stands for the ligand protonated at the N(2'). The Mössbauer spectrum from a powder sample of (I) presents a quadrupole-split doublet with parameters isomer shift (i.s.) and quadruple splitting (q.s.) values 0.126 and 0.256 mm s-1 respectively. At this stage it is worth noting the similarities of the Mössbauer parameters of Fe(II) complexes (I) and (II). Both are consistent with a low-spin iron(II) ion.
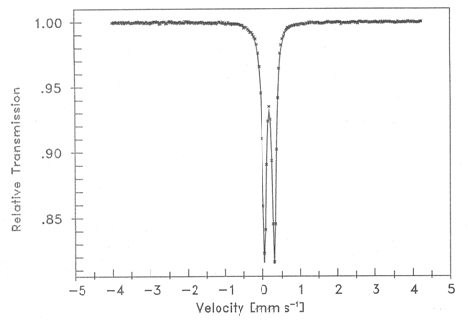 | Figure 3. Mossbauer spectrum of[ FeII(DTOH2)2]Cl2 |
3.4. Electrochemistry
- The cyclic voltammogram of the[Fe(DTOH2)2] Cl2 complex was recorded over the potential range 0.0 to 0.8 V versus silver electrode.One quasi-reversible Fe(III)/Fe(II) redox couple with a peak to peak separation of 120 mV is observed at Ef - 0.16 V (vs Fc+/Fc) , (0.44 V vs NHE) . Diacetylthiosemicarbazoneoxime donors appear to enhance the electron density at Fe(II) centers to a such an extent that Fe(II) complexes become strongly reducing. The coordination of the thione sulfur and oxime nitrogen to Fe center evidently is the major reason for the lower redox potential which is consistent with the finding that oxime ligands have a strong tendency to decrease the Mn+/(n-1)+ redox potentials of the central ions[26]. The quasi-reversible Fe(III/II) redox couple was not altered on scanning at different sweep rates, showing that the redox peak current intensity increases on higher scan rates. The cathodic peak current was found to be proportional to the square root of the scan rate indicates that the electron transfer process is diffusion controlled, which is confirmed by the observed linear plot of square root of the scan rate versus cathodic peak current.
3.5. Thermal Analysis
- The examination of TG thermo -gram reveals that the iron(II) complex follow a three – steps decomposition. The first stage shows 20.0% weight loss between 210 - 250℃, which corresponds to the loss of four CH3 radicals and two NH2 fragments (Calcd.19.4 %). The second weight loss of 15.0 % occurs in a temperature range 270 – 400℃ which corresponds to the loss of two chlorides (Calcd. 14.9 %).The third step starts at 425℃ and continues in a wider temperature range where major weight loss 50.0% occurs in a temperature range 425 – 600℃ which can be attributed to the loss of two ( NOHN-NH-CS) minus one oxygen atom (Calcd. 50.5 %). It could be inferred from the thermo-gram that the iron chelate does not contain any molecules of water.
|
ACKNOWLEDGMENTS
- We are grateful to Professor K. Wieghardt, Max-Plank Institute, for Mössbauer measurements.